Quanto vivono i bambini affetti dalla sindrome di melas? Sindrome MELAS – trattamento in Italia
La sindrome MELAS è una malattia mitocondriale caratterizzata da danni ai muscoli e al sistema nervoso centrale.
MELAS (eng. Encefalomiopatia mitocondriale, acidosi lattica ed episodi simili a ictus - "encefalomiopatia mitocondriale, acidosi lattica, episodi simili a ictus") è una malattia neurodegenerativa progressiva caratterizzata dalle manifestazioni elencate nel nome ed è accompagnata da sintomi polimorfici - ictus , diabete, convulsioni, diminuzione dell'udito, malattie cardiache, bassa statura, endocrinopatie, intolleranze attività fisica e disturbi neuropsichiatrici.
Storia.
La sindrome MELAS è stata descritta per la prima volta nel 1984 da Pavlakis e colleghi; dieci anni dopo, Pavlakis e Mizio Hirano pubblicarono una revisione di 110 casi.
Tipo di eredità:
materno
Epidemiologia:
L’esatta incidenza della malattia non è nota. In letteratura sono disponibili dati limitati sull’incidenza della malattia. Nella Finlandia settentrionale, la frequenza della mutazione A3243G è 16,3:100.000.
Patogenesi:
Le mutazioni del DNA mitocondriale, che controllano la catena respiratoria mitocondriale, sono accompagnate dall'interruzione dei processi di fosforilazione ossidativa, la fonte di energia più importante per i processi metabolici nella cellula.
Manifestazioni cliniche
All'età fino a 40 anni, i pazienti affetti da MELAS vengono ricoverati con un attacco ischemico transitorio, epilessia, vomito ripetuto, mal di testa e debolezza muscolare. A questi pazienti viene spesso diagnosticata clinicamente la demenza.
La giovane età e l’assenza di fattori di rischio caratteristici dell’ictus aiutano a pensare alla MELAS.
Dati di laboratorio
L’acidosi del lattato è un aumento dei livelli di lattato e piruvato.
Dati di visualizzazione
I cambiamenti nel cervello sono simili a quelli causati da un ictus.
Differenze dall'ictus
1) le aree interessate non coincidono con i confini dei territori vascolari arteriosi.
2) con attacchi ripetuti le lesioni vengono visualizzate in una sede diversa.
+ dati clinici (giovane età, assenza di fattori di rischio per ictus).
CT
Aree multiple ipodense non corrispondenti al territorio vascolare.
Calcificazione dei gangli della base (più comune nei pazienti anziani).
L'atrofia si verifica in un contesto di regressione e miglioramento clinico.
risonanza magnetica
Infarto miocardico acuto
Per differenziarsi da un ictus, si utilizzano ADC e DWI (con gli ictus, la diffusione è limitata (edema citotossico) e con MELAS, la diffusione è leggermente limitata o senza cambiamenti (edema vasogenico).
Coinvolgimento della sostanza bianca sottocorticale del cervello nel processo patologico.
Deterioramento nella visualizzazione della chiarezza dei contorni dei giri e aumento del segnale da essi sulle immagini pesate in T2.
Attacco cardiaco cronico
I cambiamenti possono essere simmetrici o asimmetrici.
L'atrofia focale si verifica in un contesto di regressione e miglioramento clinico.
I lobi parietali, occipitali e temporali del cervello sono più spesso colpiti.
Spettroscopia RM
Aumento dei livelli di lattato.






Parole chiave
SINDROME DI MELAS / SINDROME DI MELAS / EPILESSIA / EPILESSIA / CLINICA / QUADRO CLINICO / DIAGNOSTICA / TRATTAMENTO / TRATTAMENTOAnnotazione articolo scientifico sulla medicina clinica, autore del lavoro scientifico - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadayev V.A.
La sindrome MELAS è una malattia geneticamente determinata del gruppo delle malattie mitocondriali, definita come encefalomiopatia mitocondriale, acidosi lattica con episodi simili a ictus. Tutti gli organi e i tessuti sono coinvolti nel processo patologico, ma i sistemi muscolare e nervoso ne soffrono in misura maggiore. La malattia si sviluppa più spesso tra i 6 e i 10 anni. Il decorso della malattia è progressivo. Nella maggior parte dei casi, la malattia si manifesta con attacchi epilettici, mal di testa ricorrenti, vomito e anoressia. L’epilessia è un’importante manifestazione clinica della sindrome MELAS. Le crisi epilettiche sono il primo sintomo riconoscibile nelle encefalopatie mitocondriali (ME) nel 53% dei casi. Nella MELAS, l'epilessia occipitale è la più comune. Man mano che la malattia progredisce, l’epilessia diventa resistente alla terapia, spesso con un decorso di status. Sono stati descritti casi di trasformazione in epilessia di Kozhevnikov. Presentiamo la storia medica di un paziente con una diagnosi di sindrome MELAS verificata durante la sua vita.
Argomenti correlati lavori scientifici sulla medicina clinica, autore del lavoro scientifico - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadayev V.A.
-
Encefalopatia mitocondriale con episodi simil-ictus e acidosi lattica (sindrome di melas): criteri diagnostici, caratteristiche delle crisi epilettiche e approcci terapeutici basati su un caso clinico
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Ictus nelle malattie mitocondriali
2012 / Pizova N.V. -
Epilessia nei bambini con malattie mitocondriali: caratteristiche di diagnosi e trattamento
2012 / Zavadenko N. N., Kholin A. A. -
Disturbi neurologici nell'encefalomiopatia mitocondriale - acidosi lattica con episodi simili a ictus (sindrome MELAS)
2012 / Kharlamov Dmitry Alekseevich, Krapivkin Alexey Igorevich, Sukhorukov Vladimir Sergeevich, Kuftina Lyudmila Andreevna, Groznova Olga Sergeevna -
La sindrome di Melas come causa insolita di ipoparatiroidismo: osservazione clinica
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Belaya Zhanna Evgenievna -
Episodi simili ad ictus nell'encefalomiopatia mitocondriale con acidosi lattica
2010 / Kalashnikova Lyudmila Andreevna, Dobrynina L. A., Sakharova A. V., Chaikovskaya R. P., Mir-Kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditsky V.V., Protsky S.V. -
Citopatie mitocondriali: melas e sindromi MIDD. Un difetto genetico – diversi fenotipi clinici
2017 / Muranova A.V., Strokov I.A. -
Epilessia occipitale benigna dell'infanzia ad esordio precoce (sindrome di Panayotopoulos). Descrizione di un caso clinico
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polimorfismo delle manifestazioni cliniche dell'encefalomiopatia mitocondriale progressiva associata alla mutazione del gene POLG1
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Valore diagnostico dello studio dell'attività citochimica degli enzimi nelle malattie mitocondriali ereditarie
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
EPILESSIA NELLA SINDROME DI MELAS
La sindrome MELAS è una malattia geneticamente determinata del gruppo mitocondriale, definita come encefalomiopatia mitocondriale, acidosi lattica con episodi simili ad ictus. Il processo patologico coinvolge tutti gli organi e i tessuti, ma è per lo più dannoso per il sistema muscolare e nervoso. La malattia è più frequente nei bambini di età compresa tra 6 e 10 anni. Il decorso clinico è progressivo. Nella maggior parte dei casi la malattia si manifesta con attacchi epilettici, mal di testa ricorrenti, vomito, anoressia. L'importante presentazione clinica della sindrome MELAS è l'epilessia. Le crisi epilettiche sono il sintomo iniziale diagnosticato delle encefalopatie mitocondriali (ME) nel 53% dei casi. L'epilessia occipitale è più frequente nella sindrome MELAS. Man mano che la malattia progredisce, si osserva resistenza dell'epilessia al trattamento, spesso con comparsa di stato epilettico. Vengono descritti alcuni casi di trasformazione nell'epilessia di Kozhevnikov. Viene fornita la storia di un paziente con diagnosi verificata in vita di sindrome MELAS.
Testo del lavoro scientifico sul tema “Epilessia nella sindrome del melas”
VOLUME IV NUMERO 3 2009
EPILESSIA CON SINDROME DI MELAS
K.Yu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.B. Mikhailova2, VA. Chadayev1, A.A. Alikhanov1-2, B.N. Ryzhkov1, A.S. Petruchin1
EPILESSIA NELLA SINDROME DI MELAS
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mikhailova2, U.A. Chadaev1, A.A. Alikhanov1-2, B.N. Ryzkov1, A.S. Petruchin1
1 - Dipartimento di Neurologia e Neurochirurgia, Facoltà di Pediatria, Istituto statale di istruzione professionale superiore, Università medica statale russa di Roszdrav
2 - Stanza dei bambini russi ospedale clinico
La sindrome MELAS è una malattia geneticamente determinata del gruppo delle malattie mitocondriali, definita come encefalomiopatia mitocondriale, acidosi lattica con episodi simili a ictus. Tutti gli organi e i tessuti sono coinvolti nel processo patologico, ma i sistemi muscolare e nervoso ne soffrono in misura maggiore. La malattia si sviluppa più spesso tra i 6 e i 10 anni. Il decorso della malattia è progressivo. Nella maggior parte dei casi, la malattia si manifesta con attacchi epilettici, mal di testa ricorrenti, vomito e anoressia. L’epilessia è un’importante manifestazione clinica della sindrome MELA. Le crisi epilettiche sono il primo sintomo riconoscibile nelle encefalopatie mitocondriali (ME) nel 53% dei casi. Nella MELAS, l'epilessia occipitale è la più comune. Man mano che la malattia progredisce, l’epilessia diventa resistente alla terapia, spesso con un decorso di status. Sono stati descritti casi di trasformazione in epilessia di Kozhevnikov. Presentiamo la storia medica di un paziente con una diagnosi di sindrome MELAS verificata durante la sua vita.
Parole chiave: sindrome MELAS, epilessia, quadro clinico, diagnosi, trattamento.
La sindrome MELAS è una malattia geneticamente determinata del gruppo mitocondriale, definita come encefalomiopatia mitocondriale, acidosi lattica con episodi simili ad ictus. Il processo patologico coinvolge tutti gli organi e i tessuti, ma è per lo più dannoso per il sistema muscolare e nervoso. La malattia è più frequente nei bambini di età compresa tra 6 e 10 anni. Il decorso clinico è progressivo. Nella maggior parte dei casi la malattia si manifesta con attacchi epilettici, mal di testa ricorrenti, vomito, anoressia. L'importante presentazione clinica della sindrome MELAS è l'epilessia. Le crisi epilettiche sono il sintomo iniziale diagnosticato delle encefalopatie mitocondriali (ME) nel 53% dei casi. L'epilessia occipitale è più frequente nella sindrome MELAS. Man mano che la malattia progredisce, si osserva resistenza dell'epilessia al trattamento, spesso con comparsa di stato epilettico. Vengono descritti alcuni casi di trasformazione nell'epilessia di Kozhevnikov. Viene fornita la storia di un paziente con diagnosi verificata in vita di sindrome MELAS.
Parole chiave: sindrome MELAS, epilessia, quadro clinico, diagnostica, trattamento.
La sindrome MELAS è una malattia geneticamente determinata del gruppo delle malattie mitocondriali, definita come encefalomiopatia mitocondriale, acidosi lattica con episodi simili a ictus.
La sindrome MELAS è stata identificata per la prima volta come forma nosologica indipendente da S. Pavlakis et al. nel 1984. Tuttavia, numerosi autori suggeriscono che la malattia sia stata descritta in precedenza con il nome di “poliodistrofia familiare, miopatia mitocondriale, acidemia lattica”.
La prevalenza nella popolazione non è stata stabilita. Nel 2000 erano state pubblicate più di 120 osservazioni sulla sindrome MELAS, anche sulla stampa nazionale.
La sindrome MELAS nel 25% dei casi è ereditata per linea materna con un rischio elevato, ma nel 56-75% dei pazienti non esiste una storia familiare. La malattia è associata a mutazioni nei geni del DNA mitocondriale che codificano per subunità dei complessi della catena respiratoria e nei geni dell'RNA di trasferimento (MT-ND1, MT-ND5, MT-TH, MT-TL1 e MT-TV). Nell'80-90% dei casi di sindrome MELAS, la malattia si basa su una mutazione puntiforme nel gene MT-TL1, che codifica per l'RNA di trasferimento della leucina. Con questa mutazione, il nucleotide adenina viene sostituito dalla guanina in posizione 3243 (A3243G), che interrompe la sintesi di tutte le proteine nei mitocondri.
Tutti gli organi e i tessuti sono coinvolti nel processo patologico, ma i sistemi muscolare e nervoso ne soffrono in misura maggiore.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.B., Chadayev V.A., Alikhanov A.A., Ryzhkov B.N., Petrukhin A.S.
Epilessia nella sindrome MELAS Rus. Zhur. det. neuro.: vol. IV, fascicolo. 3, 2009.
ARTICOLI ORIGINALI
argomenti come i più dipendenti dall’energia. Pesantezza manifestazioni cliniche dipende dall'effetto soglia (età, fabbisogno energetico dei tessuti), dal controllo dei geni nucleari sulla sintesi della catena respiratoria, dall'eteroplasmia (diverso contenuto di molecole di mtDNA mutanti nei tessuti). È stato dimostrato che nei pazienti con sindrome MELAS il contenuto del mtDNA mutante in vari tessuti è del 93-96%. Nei familiari dei probandi, nei tessuti viene rilevato anche il mtDNA mutante, ma il suo contenuto è significativamente inferiore: 62-89% nella forma cancellata della malattia, dal 28 all'89% in assenza di segni clinici della sindrome.
La malattia si sviluppa più spesso tra i 6 ei 10 anni, ma ci sono casi di insorgenza precoce (fino a due anni) o successiva - da 21 a 40 anni. Prima della comparsa della malattia, il 90-100% dei pazienti si sviluppa normalmente. Il decorso della malattia è progressivo, più maligno con esordio precoce.
Nella maggior parte dei casi, la malattia si manifesta con attacchi epilettici, mal di testa ricorrenti, vomito e anoressia. Dovresti anche prestare attenzione all'intolleranza all'esercizio fisico sotto forma di deterioramento del benessere e comparsa di debolezza muscolare. Il complesso dei sintomi miopatici si manifesta con intolleranza all'esercizio, debolezza muscolare, affaticamento e talvolta ipotrofia muscolare.
Man mano che la malattia progredisce, di solito si sviluppa la demenza. Sintomi come atassia cerebellare, sordità neurosensoriale e polineuropatia periferica sono meno comuni.
Sono caratteristici gli episodi simili a ictus, che possono manifestarsi come attacchi ricorrenti di mal di testa, vertigini, sviluppo di sintomi neurologici focali (paresi, emianopsia), coma. Tali episodi acuti sono spesso precipitati da febbre o infezioni intercorrenti. Queste manifestazioni possono avere una regressione abbastanza rapida (da alcune ore a diverse settimane), così come la tendenza a ripresentarsi.
L'epilessia è un'importante manifestazione clinica che spesso si manifesta precocemente nella sindrome MELAS. Questo
spesso la manifestazione neurologica più evidente, soprattutto nelle encefalopatie mitocondriali atipiche (ME). Le crisi epilettiche sono il primo sintomo riconoscibile nelle encefalopatie mitocondriali (ME) nel 53% dei casi.
Nella MELAS, l’epilessia occipitale (OE) è il disturbo più comune. Caratteristiche sono le convulsioni focali provenienti dai lobi occipitali. Le convulsioni sono spesso associate a sintomi neurologici transitori o persistenti come la perdita del campo visivo.
Le convulsioni provenienti dalla corteccia occipitale sono suddivise in base alle loro manifestazioni in sensazioni soggettive (aura) e in sintomi clinicamente rilevabili, solitamente con una componente motoria. Le aure epilettiche emanate dal lobo occipitale comprendono allucinazioni visive semplici e complesse e amaurosi. Gli attacchi più tipici caratteristici dell'ES sono semplici allucinazioni visive, che possono manifestarsi come sintomi positivi (lampi, punti luminosi) e negativi (scotoma, emianopsia). Le allucinazioni visive più comuni sono descritte come uno o più punti luminosi, costanti o lampeggianti. Di regola, la macchia ha bianco con una tinta verdastra. Le allucinazioni possono anche essere multicolori o monocromatiche. Le allucinazioni compaiono solitamente nei campi visivi controlaterali al fuoco di eccitazione nella corteccia occipitale e successivamente si diffondono. Tuttavia, va notato che l'aura visiva non viene spesso rilevata nei reclami dei pazienti.
Allucinazioni visive complesse si osservano quando l'eccitazione epilettica si diffonde alle sezioni occipito-temporali o occipito-parietali. Le allucinazioni visive complesse possono apparire sotto forma di persone, oggetti o scene di animali, essere familiari o non familiari, piacevoli o spaventose, spaventose, semplici o grottesche, possono essere statiche o muoversi orizzontalmente e scomparire. Di norma, sono un sintomo terminale prima dello sviluppo di un attacco motorio; può essere il primo sintomo ictale, ma più spesso si verifica dopo
VOLUME IV NUMERO 3 2009
allucinazioni elementari.
Un tipo speciale, estremamente difficile da diagnosticare, di convulsioni provenienti dalla corteccia occipitale è l'ama ictale. Secondo molti autori, questo è un sintomo di irritazione del lobo occipitale tanto comune quanto le allucinazioni visive, ma spesso rimane non riconosciuto. Di solito i pazienti non identificano questo sintomo separatamente nella struttura dell'attacco. La perdita della vista avviene bilateralmente con perdita dei campi laterali. Possibile emianopsia omonima controlaterale alla fonte dell'attacco. I pazienti descrivono le sensazioni come oscuramento degli occhi, "oscurità bianca" e ridotta percezione del colore. È possibile un decorso dello stato con la formazione del cosiddetto status epilepticus amauroticus.
Le convulsioni occipitali possono presentarsi con sintomi autonomici. Questi includono emicrania, vertigini, nausea e vomito. Un sintomo comune è il mal di testa simile all’emicrania post-attacco.
Le manifestazioni cliniche delle convulsioni che si verificano limitate alla corteccia occipitale sono caratterizzate dalla deviazione degli occhi lateralmente. Si può osservare la deviazione degli occhi insieme alla deviazione della testa di lato. Nella maggior parte dei casi si verifica una deviazione degli occhi nella direzione controlaterale alla lesione. Tuttavia, sono stati descritti casi in cui si osserva una deviazione dell'occhio verso la lesione. Inoltre, una delle caratteristiche degli attacchi "occipitali" è la diffusione istantanea della scarica alle parti anteriori del cervello, mentre il quadro clinico, di regola, è dominato da una componente motoria pronunciata. Sono possibili crisi toniche, tonico-cloniche (sia emiconvulsive che secondarie generalizzate), automotorie. A questo proposito, è importante identificare i sintomi clinici iniziali: un arresto immotivato e improvviso dello sguardo, guardando oggetti inesistenti, un sorriso irragionevole, manifestazioni vegetative e una conferma necessariamente documentale della zona ittogena primaria utilizzando il metodo VEM.
Man mano che la malattia progredisce, l’epilessia diventa resistente alla terapia, spesso con un decorso di status. Sono stati descritti casi di trasformazione in epilessia di Kozhevnikov. Un certo numero di auto
dat descrive la possibilità dello stato epilettico come primo sintomo nei pazienti con MELAS senza una storia di precedenti convulsioni. Ribacoba R. et al. descrivono nella loro pubblicazione 4 casi di sviluppo di epilessia parziale continua con attacchi motori focali, preceduti da una storia di episodi di emicrania. Miyazaki M. et al. hanno mostrato la possibilità di mioclono focale continuato come parte dell'epilessia parziale continua nei pazienti con MELAS. Araki T. et al. Abbiamo osservato un paziente di 37 anni con stato epilettico di crisi focali sotto forma di fluttuazioni di coscienza, emianopsia omonima in combinazione con episodi parossistici di deviazione oculare laterale. Sull'EEG sono stati registrati modelli EEG continui di crisi localizzate nella regione occipitale. Nei pazienti adulti con MELAS vi è una predominanza di crisi motorie focali, ma l'EEG mostra una predominanza dell'attività epilettiforme multiregionale nelle regioni occipitali.
L'attività epilettiforme si registra nel 71% dei casi dopo la comparsa delle crisi. Uno studio elettroencefalografico su pazienti con sindrome MELAS è caratterizzato da attività epilettiforme nelle regioni occipitali. Numerosi autori associano la comparsa di disturbi epilettiformi regionali all'ictus. Secondo uno studio di Fujimoto S., nella fase acuta (cioè entro 5 giorni dopo un episodio simile a un ictus), la maggior parte dei pazienti esaminati con sindrome MELAS presentava onde delta regionali di elevata ampiezza in combinazione con polyspikes. Gli autori propongono di considerare questo pattern come patognomonico per episodi simili ad ictus. Oltre alle regioni occipitali, l'attività epilettiforme può diffondersi alle regioni temporali, bifrontalmente, ed anche bilateralmente alle regioni posteriori con distribuzione diffusa. Durante la fotostimolazione ritmica può verificarsi una risposta fotoparossistica.
Il principale segno di laboratorio è un aumento del livello di lattato nel sangue
ARTICOLI ORIGINALI
vi superiore a 2,0 mmol/l, che porta allo sviluppo di acidosi lattica.
risonanza magnetica del cervello fasi iniziali la malattia può essere priva di caratteristiche, anche quando si verifica l'epilessia. I metodi di neuroimaging rivelano zone infartuate negli emisferi cerebrali (80%), meno spesso nel cervelletto e nei gangli della base. Si possono osservare anche calcificazione dei gangli della base e atrofia della corteccia cerebrale. In uno studio sull'emissione di fotoni, l'accumulo dell'isotopo viene rilevato 3-16 giorni prima della comparsa della zona di infarto (diminuzione del segnale isotopico) su una scansione di tomografia computerizzata del cervello. La risonanza magnetica del cervello mostra aree di lesioni localizzate principalmente nei lobi occipitali, che possono essere transitorie. È colpita prevalentemente la corteccia occipitale, la sostanza bianca è danneggiata in misura minore. Nelle immagini pesate in T2, le lesioni cerebrali nella MELAD appaiono come aree di maggiore intensità del segnale. Numerosi autori associano aree iperintense transitorie all'edema vascolare reversibile.
L'angiografia di solito non rivela anomalie vascolari. La risonanza magnetica pesata in diffusione dimostra cambiamenti associati all'edema vasogenico.
Istopatologia: durante l'esame di una biopsia muscolare, vengono identificate fibre con "bordi rossi" frastagliati. L'autopsia cerebrale è caratterizzata da una combinazione di vecchi e nuovi focolai di infarto, nonché da atrofia corticale con focolai di necrosi.
Attualmente la terapia è di supporto. La direzione principale del trattamento è migliorare l'equilibrio energetico dei mitocondri e della catena respiratoria. Coenzima p10 (80-300 mg/giorno), vitamine K1 e K3 (25 mg/giorno), acido succinico (fino a 6 g/giorno), vitamina C (2-4 g/giorno), riboflavina (100 mg/giorno ) e nicotinamide (fino a 1 g/giorno). A causa dello sviluppo di una carenza secondaria di carnitina, ai pazienti viene prescritta L-carnitina (fino a 100 mg/kg/giorno). La vitamina E (300-500 mg/giorno) e la vitamina C (2-4 mg/giorno) sono utilizzate come terapia antiossidante.
Non esistono regimi di trattamento antiepilettico generalmente accettati per i MEDIA. Numerosi autori suggeriscono di escludere i farmaci che possono inibire il metabolismo energetico (barbiturici, farmaci a base di acido valproico, nonché alcuni farmaci di altri gruppi, ad esempio cloramfenicolo). La letteratura descrive diversi casi isolati di aggravamento di attacchi convulsivi durante l'utilizzo di acido valproico nella sindrome MELA con mutazione A3243C. I principali AED nel trattamento dell'epilessia nella sindrome MELAYA sono considerati Tegretol (o Trileptal), Topamax, Keppra a dosi terapeutiche medie. La terapia opportunamente selezionata porta ad una significativa riduzione della frequenza delle crisi generalizzate secondarie. Tuttavia, gli attacchi con compromissione delle funzioni autonomo-viscerali e visive sono generalmente resistenti al trattamento. Nella fase terminale della malattia, la frequenza delle crisi epilettiche può diminuire.
Presentiamo la storia medica di un paziente con una diagnosi verificata di sindrome MELAYA durante la sua vita.
Il paziente Ch.A., 11 anni, è stato osservato presso il Centro di Neurologia ed Epilessia Pediatrica. Al momento del ricovero, si lamentavano una graduale perdita delle capacità linguistiche, gravi disturbi dell'andatura con rifiuto di camminare, significativa diminuzione della vista, sbalzi d'umore e comportamento negativo; attacchi seriali giornalieri sotto forma di contrazioni dei muscoli facciali, dei muscoli degli arti superiori e inferiori, nonché episodi a breve termine di perdita della vista.
L'esordio della malattia è stato osservato a 5 anni e 9 mesi. Per la prima volta, in un contesto di completa salute, quando ci si addormentava si manifestava un forte mal di testa, semplici allucinazioni visive ("raggio giallo"), seguite da una rotazione forzata degli occhi e della testa di lato e dallo sviluppo di un mal di testa generalizzato attacco convulsivo tonico-clonico, dopo il quale è stato notato vomito. Dopo 9 mesi gli attacchi con gli stessi sintomi si ripresentarono e divennero rapidamente seriali. Dopo aver prescritto Tegretol alla dose di 400 mg al giorno, la frequenza degli attacchi è diminuita a 1 volta al mese. Tegretol è stato sostituito da Depakine Chrono alla dose di 900 mg/die, rispetto alla quale è stata osservata una remissione clinica per 6 mesi. Considerando i sintomi clinici
VOLUME IV NUMERO 3 2009
tomatismo, tempistica degli attacchi durante il periodo di addormentamento, intelligenza normale del paziente, reazione positiva al valproato, epilessia occipitale idiopatica.
All'età di 7 anni, le crisi focali versive riprendono con generalizzazione secondaria dopo essersi addormentati con la stessa frequenza di 1 volta al mese. L'aumento della dose di depakin a 1500 mg/die non ha portato ad una diminuzione della frequenza degli attacchi. Con l'aggiunta di Lamictal alla dose di 75 mg/die gli attacchi si sono interrotti per 4 mesi, per poi riprendere con la stessa frequenza. All'età di 8 anni sono iniziati gli attacchi con perdita della vista a breve termine. Da 8 anni 8 mesi. prima di addormentarsi cominciavano a manifestarsi assenze atipiche: ammiccamento rapido con chiusura delle palpebre e spostamento dei bulbi oculari verso l'alto; la coscienza fluttua.
All'età di 9 anni si manifestavano attacchi seriali multipli, della durata di diversi giorni, con semplici allucinazioni visive sotto forma di un "raggio" che lampeggiava davanti agli occhi, con gli occhi e la testa girati a destra. Prima di addormentarsi, tali attacchi a volte si trasformavano in focali emiclonici, che si manifestavano con la contrazione del viso
muscoli a destra, spasmo della testa a destra, clonazione degli arti destri (più di un braccio). A volte dopo un attacco si verificavano forti mal di testa e vomito. Alla stessa età apparvero attacchi inibitori: un'aura sotto forma di sensazione di pelle d'oca pollice piede destro, seguito da debolezza transitoria della gamba destra e goffaggine del braccio destro. Topamax è stato introdotto nel regime di trattamento alla dose di 100 mg/giorno e per 1 anno non si sono verificati attacchi epilettici.
Inoltre, all'età di 9 anni, sono comparse per la prima volta condizioni parossistiche, accompagnate da forte mal di testa, vomito e sviluppo di emiparesi del lato destro. In alcuni casi, tali condizioni erano accompagnate da amaurosi che durava da alcuni minuti a diversi giorni.
All'età di 10,5 anni, gli attacchi riapparivano sotto forma di rotazione della testa a sinistra, movimenti a scatti dei bulbi oculari verso sinistra, che duravano fino a 5 secondi, con una frequenza fino a 3 volte all'ora, al giorno, anche durante sonno. La dose di Topamax è stata aumentata a 150 mg/die senza effetti significativi. A 10 anni e 10 mesi. dopo un forte mal di testa, alterna-
Riso. 1. Paziente Ch.A. 10 anni. Diagnosi: sindrome MEAE. Epilessia focale sintomatica.
Monitoraggio video-EEG (2004): sullo sfondo di un diffuso rallentamento dell'attività cerebrale principale, si registra una continua attività epilettiforme nella regione occipitale sinistra. Sono stati registrati anche modelli EEG subclinici di crisi epilettiche nella regione occipitale sinistra, con diffusione alla regione temporale posteriore sinistra.
Centro di Neurologia ed Epilessia Pediatrica
sotto la guida del Professor K.Yu. Mukhina è impegnata nella diagnosi e nel trattamento delle malattie del sistema nervoso ad Aetey, specializzata nelle forme di epilessia etiana.
Direzioni principali
attività:
Epilessia nei bambini e negli adolescenti
Disturbi del sonno nei bambini
Tic, enuresi
Esame dei bambini nei primi mesi di vita.
Esami nel nostro centro:
Diagnosi e trattamento delle malattie del sistema nervoso nei bambini
Diagnostica completa (inclusa quella prechirurgica) e trattamento dell'epilessia
Consultazione con neurologi ed epilettologi
Consultazione con un pediatra (bambini frequentemente malati, gastroenterologia, ecc.)
Consultazione con uno psichiatra e uno psicologo.
Consultazione con un genetista con test (compreso il cariotipo)
Monitoraggio video-EEG (nelle sale appositamente attrezzate del Centro o visitando il domicilio del paziente)
Elettroencefalografia computerizzata (digitale).
Dopplerografia ad ultrasuoni (ecografia Doppler) dei vasi della testa e del collo
Ecoencefalografia (ECHO EG)
Sul nostro sito web è possibile abbonarsi alla rivista “Russian Journal of Child Neurology” tramite Internet.
Informazioni dettagliate sul lavoro del Centro dalle 10:00 alle 19:00 telefonicamente:
Tel.: (+7495)983-09-03; (+7926)290-50-30 Tel./fax: (+7495) 394-82-52
Indirizzo: S. Stagni Borisovskie, 13, edificio. 2. Internet: www.epilettologo.ru E-mail: [e-mail protetta](per indicazioni dettagliate consultare il sito)
VOLUME IV NUMERO 3 2009
crisi epilettiche focali emcloniche e generalizzate secondarie, che sono diventate seriali e sono durate 48 ore. Frisium è stato aggiunto a Topamax alla dose di 10 mg/die con un effetto positivo temporaneo.
Dall'età di 8 anni si cominciarono a notare difficoltà nell'apprendimento del materiale scolastico; la memoria è diminuita. Sono comparsi aumento della fatica, dell'esaurimento e dell'inibizione dell'attività mentale. Il ragazzo divenne lunatico, irritabile e negativo; l'atmosfera di fondo è diminuita. Dall'età di 9 anni è stato notato un aumento di questi sintomi.
Dall'anamnesi si sa che il bambino è nato da una seconda gravidanza normale, un parto al secondo termine, peso alla nascita 2800 g, lunghezza 53 cm. Lo sviluppo psicomotorio e del linguaggio precoce era pienamente coerente con l'età. Malattie precedenti: varicella all'età di 6 anni, ARVI frequente (fino a 4 volte l'anno) dall'età di 6 anni. L'ereditarietà per l'epilessia e altre malattie neurologiche non è gravata.
Al momento della visita (11 anni), le condizioni del bambino erano gravi; reagisce negativamente all'ispezione. Consapevole, orientato alla pro-
viaggio e tempo. È estremamente riluttante a stabilire un contatto e si rifiuta di seguire le istruzioni. Nistagmo spontaneo a sinistra, la testa è inclinata verso la spalla sinistra con una rotazione a destra. La lingua è sulla linea mediana, il riflesso faringeo è ridotto; Si notano disfagia e disartria. La vista è ridotta.
Si determina un'ipotonia muscolare diffusa moderata. I riflessi tendinei sono uniformemente ridotti. Si è verificata una leggera diminuzione della forza muscolare nelle estremità destre. Non sono stati rilevati riflessi patologici del piede. Non ci sono prove oggettive di compromissione sensoriale. Non c'è Romberg nel campione. Si rifiuta di camminare. Quando cerca di rimetterlo in piedi, piange e si siede sul pavimento. Mancante quando si esegue un test dell'indice del dito. Parla lentamente, con parole separate, con riluttanza.
Metodi aggiuntivi esami. Monitoraggio video-EEG (2004). Rallentamento significativo dell'attività principale di registrazione in background. Nel corso dello studio è stata registrata una continua attività epilettiforme nella regione occipitale sinistra, con estensione alla regione temporale posteriore sinistra e con la formazione periodica di un pattern EEG.
Nato nel 1993 16/12/05
Riso. 2. Paziente Ch.A. 11 anni. Diagnosi: sindrome MELAS. Epilessia focale sintomatica.
Il monitoraggio video-EEG è stato effettuato nel tempo dopo 1 anno (2005): significativo rallentamento dell'attività cerebrale di fondo. Durante la registrazione del sonno, viene registrato un rallentamento regionale continuo nella regione fronto-centrale destra, la cui struttura rivela l'attività delle onde di picco nella regione fronto-centrale destra.
ARTICOLI ORIGINALI
stupa (Fig. 1). Un rallentamento regionale continuo viene rilevato anche nella regione frontale-centrale destra con l'inclusione di singole onde acute.
Monitoraggio video-EEG in dinamica (2005): rallentamento significativo dell'attività cerebrale di fondo. Durante lo studio, è stato registrato un continuo rallentamento regionale nella regione frontocentrale destra. Nella struttura del rallentamento regionale nella regione frontale-centrale destra, viene rilevata l'attività delle onde di picco (Fig. 2).
risonanza magnetica del cervello. La prima risonanza magnetica (6 anni) ha rivelato un singolo segnale iperintenso in modalità T2 nell'emisfero sinistro del cervelletto. Studio MRI nel tempo (10,5 anni): è stato rivelato un significativo deterioramento della lesione primaria con l'estensione del processo patologico ampiamente alle regioni occipitale-parietali sinistra e destra di entrambi gli emisferi del cervello (professor A.A. Alikhanov).
Potenziali evocati visivi: cambiamenti morfofunzionali significativi nel sistema afferente visivo a livello del nervo ottico e della parte corticale dell'analizzatore visivo, più pronunciati a sinistra.
Consultazione con un oculista: atrofia parziale dei nervi ottici. Elementi di agnosia corticale.
Elettrocardiogramma: ritmo ectopico con accelerazione fino a 100 battiti al minuto.
Posizione verticale dell'asse elettrico del cuore. Cambiamenti nei processi di ripolarizzazione, che sono più pronunciati nell'ortostasi.
Elettroneuromiografia: è stata rilevata una lesione di tipo muscolare primario. Le velocità di conduzione lungo i nervi periferici non sono ridotte.
Studio del livello di lattato nel sangue: il livello di lattato nel sangue è 3,0 mmol/l (il valore normale è fino a 1,8).
Considerando la presenza di crisi epilettiche provenienti dalle parti occipitali della corteccia cerebrale, resistenti alla terapia, episodi simili a ictus, periodi di amaurosi, diminuzione delle funzioni cognitive, presenza di segnali iperintensi alla risonanza magnetica nel cervelletto e nelle parti posteriori della corteccia cerebrale , aumento dei livelli di lattato nel sangue, al paziente è stata suggerita la diagnosi di sindrome MELAS. Durante un esame genetico, la mutazione A3243G nello stato eteroplasmico è stata rilevata nelle cellule del sangue (la diagnosi è stata effettuata presso il Centro di ricerca statale di Mosca dell'Accademia russa delle scienze mediche) e la diagnosi è stata verificata.
L'osservazione successiva ha mostrato una rapida progressione dei disturbi delle funzioni mentali superiori, sviluppo di cecità corticale, completa immobilità del paziente, seguita dalla morte all'età di 12 anni e 10 mesi. (7 anni dopo l'esordio della malattia).
Bibliografia
1. Nikolaeva E.A., Temin P.A. Malattie mitocondriali accompagnate da disturbi dello sviluppo neuropsichico. Sindrome MELAS // Disturbi ereditari dello sviluppo neuropsichico dei bambini. Una guida per i medici a cura di Temin P.A. Kazantseva L.Z. - Medicina, 2001. - P. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Trattamento di un bambino con sindrome mitocondriale MELAS (encefalopatia mitocondriale, acidosi lattica, episodi simili a ictus) // Bollettino russo di perinatologia e pediatria. - 1997. - N. 2. - P. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina Z. Decorso simile all'ictus dell'encefalomiopatia mitocondriale (sindrome MELAS) // Atmosfera. Malattie nervose. - 2006. - N. 1. - pp. 43-48.
4. Temin P.A., Nikanorova M.Yu., Nikolaeva E.A. Sindrome MELAS (encefalomiopatia mitocondriale, acidosi lattica, episodi simili a ictus): principali manifestazioni, criteri diagnostici, opzioni terapeutiche // Neurol. rivista - 1998. - N. 2. - pp. 43-48.
5. Ajmone-Marsan C., Ralston B. La crisi epilettica, la sua morfologia funzionale e significato diagnostico. - Springfield (IL): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Cecità corticale ictale con perdita permanente della vista // Epilessia. - 1989. - V. 30. - P. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Un caso di MELAS che presenta uno stato epilettico parziale complesso // Rinsho Shinkeigaku. - 2001. - V.41(8). - P. 487-90.
VOLUME IV NUMERO 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic fenotipi associati a disturbi mitocondriali // Neurologia. - 2001. - V.56(10). - P. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. L'acido valproico aggrava l'epilessia dovuta alla MELAS in un paziente con una mutazione A3243G del DNA mitocondriale // Metab Brain Dis. - 2007 - V.22(1). - P. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Patologia molecolare di MELAS e MERRF. La relazione tra carico di mutazioni e fenotipi clinici // Cervello. - 1997. -V.120. - P. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polimorfismo dell'epilessia associato alla mutazione A3243G del DNA mitocondriale (MELAS): ragioni per una diagnosi ritardata // Rev Neurol (Parigi). - 2004. - V. 160(8-9). - P. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Emicrania classica, epilessia intrattabile e ictus multipli: una sindrome correlata all'encefalopatia mitocondriale / In: Andermann F., Lugaresi E., editori. Emicrania ed epilessia. - Boston: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Risultati elettroencefalografici seriali nei pazienti con MELAS // Pediatr Neurol. - 1999. - V.20(1). - P. 43-48.
14. Goto Y., Nonaka I., Horai S.A. Una mutazione nel gene tRNA leu(UUR) associato al sottogruppo MELAS delle encefalomiopatie mitocondriali // Natura. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Tomografia computerizzata e angiografia nella MELAS (miopatia mitocondriale, encefalopatia, acidosi lattica ed episodi simil-ictus): descrizione di 3 casi // Neuroradiologia. - 1987. -V. 29. - P. 393-397.
16. Hirano M., Pavlakis S.G. Miopatia mitocondriale, encefalopatia, acidosi lattica ed episodi simili a ictus (MELAS): concetti attuali // J. clin. Neurolo. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Crisi epilettiche in un paziente con miopatia mitocondriale, encefalopatia, acidosi lattica ed episodi simili a ictus ( MELAS) // Jpn J Psichiatria Neurol. - 1989. - V.43(3). - P. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Encefalomiopatia mitocondriale con aumento del lattato-piruvato e infarti cerebrali // Neurologia. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Epilessia sintomatica del lobo occipitale // Epilessia. - 1998. - V. 39 Supplemento 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Profondità e registrazione corticale diretta nei disturbi convulsivi di origine extratemporale // Neurologia. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Modelli ictali clinici in pazienti epilettici con focolai elettroencefalografici occipitali // Neurologia. - 1975. - V. 25. - P. 463-471.
22. Matthews P. M., Tampieri D., Berkovic S. F., Andermann F., Silver K., Chityat D., et al. La risonanza magnetica mostra anomalie specifiche nella sindrome MELAS // Neurologia. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Un caso con MELAS associato a epilessia parziale continua // No To Hattatsu. - 1991. - V.23(1). - Pag. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. Sindrome MELAS: caratteristiche emicraniche ed epilettiche e trasmissione materna // Neurologia. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Flusso sanguigno cerebrale nella miopatia mitocondriale, encefalopatia, acidosi lattica ed episodi simili a ictus // Ictus. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Miopatia mitocondriale, encefalopatia, acidosi lattica ed episodi simili a ictus: una sindrome clinica distintiva // Un neurolo. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Caratteristiche dello stato epilettico nel MELAS. Analisi di quattro casi // Neurologia. - 2006. - V.21(1). - P. 1-11.
28. Williamson P.D., Spencer S.S. Caratteristiche cliniche ed EEG delle crisi parziali complesse di origine extratemporale // Epilessia. - 1986. - V. 27 (Suppl 2). - P. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Epilessia del lobo occipitale: caratteristiche cliniche, modelli di diffusione delle crisi e risultati dell'intervento chirurgico // Ann Neurol. - 1992. - V. 31. - P. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Effetto paradosso del valproato di sodio che aggrava l'epilessia della MELAS in un paziente con mutazione A3243G del DNA mitocondriale // Central European Journal of Medicine. - 2007. - V.2(1). - P.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Edema vasogenico su MELAS: uno studio seriale con neurologia pesata in diffusione. - 1999. - V. 53. - P. 2182-2184.
Sindrome MELAS(dall'inglese Mitochondrial Encephalomyapathy, Lactic Acidosis, and Stroke-like Episodes) - encefalomiopatia mitocondriale, acidosi lattica ed episodi simili a ictus - si manifesta solitamente intorno ai 5-20 anni di età. La malattia si manifesta principalmente con episodi acuti simili a ictus con lo sviluppo di alterazioni focali nelle regioni occipitale e parieto-temporale del cervello e la comparsa di corrispondenti sintomi neurologici (paresi, disturbi visivi corticali, convulsioni, coma, attacchi di mal di testa e vomito, ecc.). La comparsa di lesioni è associata a disfunzione transitoria della fosforilazione ossidativa nel parenchima cerebrale, nonché a disturbi strutturali e metabolici nelle pareti delle arteriole e dei capillari; tratto caratteristico Tali infarti cerebrali “misti” sono caratterizzati da un recupero relativamente rapido.
Per la sindrome MELAS Si possono osservare anche manifestazioni miopatiche (aumento della fatica e dell'intolleranza all'esercizio fisico), depressione, atassia, degenerazione retinica, sordità neurosensoriale, bassa statura, diabete, cardiomiopatia e una serie di altre manifestazioni multiorgano. È caratteristico un livello significativo di acidosi lattica nel sangue e nel liquido cerebrospinale. Le biopsie dei muscoli scheletrici spesso rivelano il fenomeno delle "fibre rosse strappate". La sindrome MELAS è ereditata dalla madre, ma l'estrema variabilità delle manifestazioni cliniche può rendere molto difficile la valutazione della storia familiare.
Nei pazienti con sindrome MELAS Sono state descritte almeno 8 mutazioni puntiformi nei geni del mtDNA e 5 di esse sono localizzate in parti diverse del gene del tRNA). La mutazione più comune è la sostituzione A->G nella posizione 3243 (circa l'80% dei pazienti) e, in generale, mutazioni in questo gene del tRNA della leucina si riscontrano in quasi il 95% dei casi MELAS. In rari casi, in pazienti affetti da MELAS sono state descritte mutazioni puntiformi nei geni di altri tRNA e nel gene COX della subunità III del complesso IV della catena respiratoria. Tutte le mutazioni si trovano nello stato eteroplasmico.
Sindrome NARP(dall'inglese Neuropathy, Atax, Retinitis Pigmentosa) - neuropatia con atassia e retinite pigmentosa - è caratterizzata, secondo il nome, dallo sviluppo di neuropatia periferica progressiva con debolezza muscolare, atassia cerebellare e degenerazione pigmentaria retinica. Come per le altre encefalomiopatie mitocondriali, il quadro clinico può essere molto variabile, con la presenza o l'assenza di numerosi sintomi aggiuntivi nei parenti (ritardo dello sviluppo psicomotorio, crisi epilettiche, demenza). I test per l'acidosi lattica e altri marcatori di disfunzione mitocondriale non sono sempre informativi.
Tipo di ereditarietà della malattia materno. Tutti i pazienti affetti dalla sindrome NARP presentano una mutazione eteroplasmica T=>G in posizione 8993 (gene ATPasi 6 - subunità V del complesso della catena respiratoria). Il livello di eteroplasmia è decisivo per la natura della manifestazione di questa mutazione: quando contiene mtDNA mutante<78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея .
 Indice dell'argomento "Patologia mitocondriale del sistema nervoso":
Indice dell'argomento "Patologia mitocondriale del sistema nervoso": La sindrome MELAS si riferisce alle malattie mitocondriali (MD), che sono causate da difetti genetici e biochimici strutturali dei mitocondri e sono accompagnate da una respirazione alterata dei tessuti e, di conseguenza, da un difetto sistemico nel metabolismo energetico, a seguito del quale la maggior parte dell'energia I tessuti dipendenti e gli organi bersaglio sono colpiti in varie combinazioni: cervello, muscoli scheletrici e miocardio, pancreas, organo della vista, reni, fegato. I disturbi clinici in questi organi possono verificarsi a qualsiasi età. Allo stesso tempo, l’eterogeneità dei sintomi complica la diagnosi clinica di queste malattie. La necessità di escludere il MB nasce in presenza di manifestazioni multisistemiche che non rientrano nel consueto processo patologico. La frequenza delle disfunzioni della catena respiratoria è stimata da 1 su 5 - 10mila a 4 - 5 su 100mila neonati.
leggi anche il post: Malattie mitocondriali(al sito web)
La sindrome MELAS (Encefalomiopatia Mitocondriale, Acidosi Lattica ed Episodi Simil-Ictus) è una malattia multisistemica caratterizzata da episodi simil-ictus che si manifestano in giovane età (prima dei 40 anni), encefalopatia con convulsioni e demenza, miopatia mitocondriale con il fenomeno dell'” fibre rosse “stracciate” e acidosi lattica (è possibile aumentare il livello di acido lattico nel sangue senza acidosi).
La sindrome MELAS è causata da mutazioni puntiformi nel DNA mitocondriale (mtDNA). La malattia è ereditata per parte materna (quindi, i parenti da parte materna sono probabili portatori di tali mutazioni; più spesso, i parenti da parte materna descrivono un quadro clinico oligosintomatico con sintomi individuali della sindrome MELAS; nei parenti con decorso asintomatico, MELAS la sindrome viene identificata solo dai risultati della biopsia muscolare o della ricerca molecolare). Attualmente sono noti più di dieci geni le cui mutazioni portano allo sviluppo del quadro clinico della sindrome MELAS. Nella maggior parte dei casi, lo sviluppo della sindrome MELAS è causato da mutazioni nei geni che codificano le funzioni dell'RNA di trasferimento.
Tipicamente, la malattia esordisce tra i 6 e i 10 anni (l'età di esordio varia dai 3 ai 40 anni; l'esordio precoce della malattia è tipico e si verifica nel 90% dei pazienti). I pazienti sono caratterizzati da bassa statura (e intolleranza all'esercizio). Da parte degli organi interni si possono osservare cardiomiopatia, disturbi della conduzione cardiaca, diabete mellito, nefropatia e ridotta motilità del tratto gastrointestinale.
Ricordare! 1 I principali criteri clinici per la diagnosi di MELAS sono: [ 2 ] tipo di eredità materna; [ 3 ] esordio prima dei 40 anni; [ 4 ] normale sviluppo psicomotorio prima della malattia; [ 5 ] intolleranza all'esercizio fisico; [ 6 ] mal di testa simile all'emicrania con nausea e vomito; [ 7 ] episodi simili ad ictus; [ 8 ] encefalopatia con crisi epilettiche e/o demenza (le crisi miocloniche sono più spesso registrate, ma si notano anche crisi focali sensoriali, motorie e tonico-cloniche generalizzate secondarie); [ 9 ] acidosi lattica; [ 10 ] fibre rosse sfilacciate nelle biopsie dei muscoli scheletrici; [
] andamento progressivo. !!! ] molto spesso le lesioni sono localizzate nella corteccia dei lobi occipitali degli emisferi cerebrali, il che porta all'emianopsia o cecità corticale). Gli ictus possono risolversi o essere determinati a lungo termine sotto forma di cambiamenti clinici e/o radiologici (che dipendono dalla gravità dei disturbi metabolici causati dalla carenza energetica dei neuroni). Spesso "infarti cerebrali" ripetuti si sviluppano ad intervalli da 1 a 3 mesi in aree simmetriche. Queste lesioni possono essere piccole o grandi, singole o multiple, solitamente sono asimmetriche e la loro localizzazione non corrisponde all'area di irrorazione sanguigna. Inoltre, i pazienti con sindrome MELAS possono presentare calcificazioni nei gangli della base (in questi casi può essere utile una TC del cervello). Nello stato neurologico, questi cambiamenti morfologici si manifestano con mioclono, atassia, episodi di psicosi acuta o disturbi (deficit) della coscienza fino al coma ([ !!! ] caratteristica di questi episodi acuti, incl. ictus, da un lato, una rapida [da alcune ore a diverse settimane] regressione dei sintomi, dall'altro, una tendenza alla ricaduta); dagli organi di senso, vengono rilevati atrofia dei nervi ottici, retinopatia pigmentaria e perdita dell'udito.
Si ritiene che i seguenti meccanismi siano importanti nella genesi dell’IPE: [ 1 ] disturbi metabolici nel cervello con sviluppo di acidosi lattica dovuta a carenza di energia mitocondriale; [ 2 ] ischemia cerebrale causata da angiopatia mitocondriale a livello delle piccole arterie; [ 3 ] aumento locale dell'eccitabilità neuronale dovuto alla disfunzione mitocondriale nei neuroni, negli astrociti o nell'endotelio capillare, che si diffonde gradualmente in tutta la corteccia cerebrale, si combina con lo sviluppo di edema e può portare alla necrosi laminare nella corteccia cerebrale.
A uno sguardo superficiale, un ictus con sindrome MELAS è simile a un ictus normale dovuto a trombosi o embolia. In effetti, gli episodi di tipo ictus nella sindrome MELAS sono atipici: si verificano nei giovani, sono spesso provocati da malattie infettive e possono presentarsi sotto forma di mal di testa o convulsioni simili all'emicrania. La scansione MRI dell'IPE acuto nella sindrome MELAS rivela anomalie come un aumento del segnale sulle immagini pesate in T2 o FLAIR (recupero dell'inversione attenuato dall'acqua). Le lesioni non coincidono con i territori delle principali arterie cerebrali, ma coinvolgono largamente la corteccia e la sostanza bianca sottostante, con un moderato coinvolgimento della sostanza bianca profonda. Le lesioni cerebrali acute alla risonanza magnetica nella sindrome MELAS possono cambiare, migrare o addirittura scomparire ([ !!! ] caratterizzato dalla fluttuazione dei fuochi, determinata mediante MRI). L'angiografia rivela l'assenza di patologie vascolari significative: oltre ai risultati normali si può rilevare un aumento del calibro delle arterie, delle vene o un'iperemia capillare.
Gli studi neuromorfologici del cervello nella sindrome MELAS mostrano la presenza di necrosi multifocale, localizzata principalmente nella corteccia cerebrale e nella sostanza bianca sottocorticale, nonché nel cervelletto, nel talamo e nei gangli della base. Le lesioni ricordano aree di infarto, ma, come accennato in precedenza, non coincidono con i bacini dei grandi vasi cerebrali. C'è anche la degenerazione spongiforme della corteccia cerebrale, la proliferazione dei capillari e l'esaurimento dei neuroni.
Ricordare! 1 Gli episodi simili ad ictus in MELAS hanno le seguenti caratteristiche: [ 2 ] giovane età (di solito fino a 40 anni); [ 3 ] presenza frequente di un fattore provocante (si verifica dopo febbre febbrile, attacco epilettico, mal di testa simile all'emicrania); [ 4 ] localizzazione preferita - regione occipitale; [
] le lesioni, di regola, si trovano al di fuori dell'area delle grandi arterie cerebrali, il più delle volte situate nella corteccia o nelle strutture profonde della sostanza bianca del cervello.
Quando si effettua una diagnosi differenziale di IPE e infarto cerebrale, vengono presi in considerazione i seguenti sintomi:
■ una diminuzione graduale del livello di veglia, che è in dissonanza con un deficit neurologico focale relativamente lieve, non è accompagnata da una sindrome secondaria del tronco encefalico e, pertanto, non può essere spiegata da un aumento dell'infarto cerebrale e dell'edema (questi sintomi sono anche basato su un disturbo metabolico causato da una violazione dell'approvvigionamento energetico del cervello);
■ sviluppo nel periodo acuto di ripetute crisi epilettiche locali e generalizzate, che, secondo la letteratura, si verificano in 2/3 dei pazienti con IPE (le crisi non sono associate ad accidente cerebrovascolare, poiché la fonte della loro generazione è l'attività di scarica in entrambi emisferi cerebrali, e non in strutture confinate in un bacino specifico di arterie cerebrali, anche la natura ricorrente delle convulsioni e l'assenza di sintomi neurologici focali persistenti pronunciati non sono caratteristici dell'infarto cerebrale acuto);
■ completa pervietà delle arterie cerebrali secondo la scansione duplex delle arterie brachiocefaliche e l'angiografia cerebrale, che non è tipica dell'ictus ischemico;
■ caratteristiche del quadro neuroimaging: localizzazione prevalentemente corticale dei focolai e la loro “localizzazione posteriore”, che è caratteristica della MELAS e si spiega con la maggiore vulnerabilità dei neuroni in queste aree a causa del loro maggiore fabbisogno energetico; Un'altra caratteristica del neuroimaging è la scomparsa di alcuni focolai, che, a quanto pare, si basa sull'edema e non sulla necrosi della sostanza cerebrale dovuta a disturbi metabolici.
![]()
Sindrome MELAS SU RADIOPEDIA.org
Una delle principali manifestazioni della sindrome MELAS è anche la debolezza muscolare (sindrome miopatica). Tuttavia, la non specificità di questo sintomo non consente una diagnosi. Solo quando si verificano emicrania, convulsioni e/o eventi simili a ictus è possibile diagnosticare l'esordio della sindrome MELAS.
I test di screening per la sindrome MELAS sono il neuroimaging e uno studio del livello [aumento] di lattato nel sangue (parzialmente nel liquido cerebrospinale) - un esame del sangue per l'acido lattico (lattato) e piruvico (livello di lattato nel sangue [normale] - sangue venoso - 0,5 - 2,2 mmol/l, sangue arterioso - 0,5 - 1,6 mmol/l; rapporto lattato/piruvato - 10/1). La diagnosi può essere confermata dal test del DNA per determinare le mutazioni più comuni. In assenza di mutazioni puntiformi comuni nella sindrome MELAS, una biopsia muscolare (utilizzando il metodo a tre colori di Gomori per rilevare le fibre rosse sfilacciate [RRF] - miofibrille con un alto contenuto del genoma mutante e un gran numero di mitocondri alterati proliferanti) può aiuto nella diagnosi. Essa (biopsia) consente inoltre di determinare la presenza di difetti biochimici nella catena respiratoria, principalmente associati agli enzimi succinato deidrogenasi e citocromo ossidasi.

Il trattamento della sindrome MELAS comprende due aree principali. La prima è la terapia sindromica (il focus è su epilessia, diabete, ecc.). Non differisce dagli approcci generalmente accettati al trattamento delle sindromi. È necessario fermare le crisi epilettiche perché lo stress metabolico che si verifica durante le crisi può innescare lo sviluppo di episodi simili a ictus. I derivati dell'acido valproico, ampiamente utilizzati in epilettologia, inibiscono le funzioni mitocondriali e il loro utilizzo è indesiderabile. Se è impossibile interrompere il farmaco, si deve assumere contemporaneamente levocarnitina ad una dose massima di 100 mg/kg al giorno. Anche la fenitoina e i barbiturici dovrebbero essere evitati. La seconda direzione del trattamento è patogenetica, ma attualmente non esiste una terapia patogenetica efficace. La strategia di trattamento è mirata a migliorare il metabolismo energetico della cellula e comprende la somministrazione di coenzima Q o idebinone (Noben), preparati di acido succinico, vitamine K1 e K3, nicotinamide, riboflavina, L-carnitina, antiossidanti (mexidol, Mildronato, vitamine E e C), correttori del lattato-acidosi (dimefosfone). [ !!!
] È necessario evitare l'uso di farmaci che inibiscono la funzione mitocondriale (barbiturici, valproati, statine, glucocorticoidi).
Maggiori informazioni sulla sindrome MELAS nelle seguenti fonti:
presentazione “Sindrome MELAS” Kuzenkova L.M., Globa O.V.; Dipartimento di Psiconeurologia, Istituto di ricerca di pediatria, Centro scientifico per la salute dei bambini, Accademia russa delle scienze mediche, Mosca [leggi];
articolo “Encefalopatia mitocondriale con episodi simili a ictus e acidosi lattica (sindrome MELAS): criteri diagnostici, caratteristiche delle crisi epilettiche e approcci al trattamento utilizzando l'esempio di un caso clinico” Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A.; Istituzione statale autonoma della regione di Rostov “Centro diagnostico e consultivo regionale”; Dipartimento di Neurologia e Neurochirurgia con corsi di terapia manuale e riflessologia della Facoltà di Educazione e Formazione dell'Istituto di istruzione superiore di bilancio dello Stato federale "Università medica statale di Rostov" del Ministero della sanità russo (rivista "Neurologia, neuropsichiatria, psicosomatica " N. 9(4), 2017) [leggi];
articolo “Citopatie mitocondriali: sindromi MELAS e MIDD. Un difetto genetico – diversi fenotipi clinici” Muranova A.V., Strokov I.A.; Istituto di istruzione superiore di bilancio dello Stato federale “Prima università medica statale di Mosca che porta il nome. LORO. Sechenov" Ministero della Salute della Federazione Russa, Mosca (Giornale neurologico, n. 1, 2017) [leggi];
articolo “Episodi simili a ictus nell’encefalomiopatia mitocondriale con acidosi lattica” L.A. Kalashnikova, Los Angeles Dobrynina, A.V. Sakharova, R.P. Čajkovskaja, M.F. Mir-Kasimov, R.N. Konovalov, A.A. Shabalina, M.V. Kostyreva, V.V. Gnezditsky, S.V. Protskij; Centro Scientifico di Neurologia dell'Accademia Russa delle Scienze Mediche, Mosca (rivista “Annali di Neurologia Clinica e Sperimentale” n. 3, 2010) [leggi];
articolo “Disturbi neurologici nell'encefalomiopatia mitocondriale - acidosi lattica con episodi simili ad ictus (sindrome MELAS)” D.A. Kharlamov, A.I. Krapivkin, V.S. Sukhorukov, L.A. Kuftina, O.S. Groznova; Istituto di ricerca di pediatria e chirurgia pediatrica di Mosca (rivista “Bollettino russo di perinatologia e pediatria” n. 4(2), 2012) [leggi];
articolo “Decorso simile all'ictus dell'encefalomiopatia mitocondriale (sindrome MELAS)” di I.N. Smirnova, BA Kistenev, M.V. Krotenkova, Z.A. Suslina; Centro scientifico di neurologia dell'Accademia russa delle scienze mediche, Mosca (rivista “Malattie nervose” n. 1, 2006) [leggi];
articolo “Ictus nelle malattie mitocondriali” N.V. Pizova, Dipartimento di malattie nervose con corsi di neurochirurgia e genetica medica, Accademia medica statale di Yaroslavl (rivista “Neurologia, neuropsichiatria, psicosomatica” n. 9(4), 2017) [leggi];
articolo “Ictus ischemico come manifestazione di encefalopatia mitocondriale in un paziente giovane” Murzaliev A.M., Lutsenko I.L., Musabekova T.O., Akbalaeva B.A. (rivista “Scienza e Nuove Tecnologie” n. 6, 2011) [leggi];
articolo “Epilessia nella sindrome MELAS” Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadayev V.A., Alikhanov A.A., Ryzhkov B.N., Petrukhin A.S.; Istituto statale di istruzione professionale superiore RSMU Roszdrav; Ospedale clinico pediatrico russo (Giornale russo di neurologia infantile" n. 3, 2009) [leggi];
articolo “Algoritmo per la diagnosi delle encefalomiopatie mitocondriali” di S.N. Ilarioshkin, Istituto di ricerca di neurologia dell'Accademia russa delle scienze mediche (rivista "Malattie nervose" n. 3, 2007) [leggi]
©Laesus De Liro
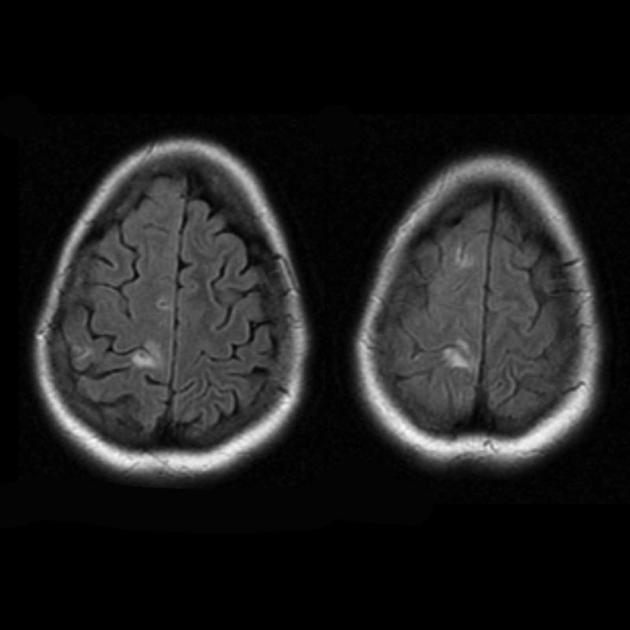
MELAS (encefalomiopatia mitocondriale con acidosi lattica ed episodi simili ad ictus, Inglese MELAS) è una malattia appartenente al gruppo delle malattie mitocondriali. I mitocondri sono organelli dotati di un proprio DNA, che viene trasmesso ed ereditato attraverso la linea materna. I tipici cambiamenti nell'imaging includono focolai simili a ictus a vari stadi di sviluppo (modello "shifting spread") che non coincidono con i confini dei letti vascolari arteriosi, e c'è una certa predisposizione alla comparsa di lesioni nelle parti posteriori del lobi parietali e occipitali. La spettroscopia MR può dimostrare un picco elevato del lattato anche quando il cervello è normale.
Epidemiologia
La sindrome MELAS si presenta solitamente come episodi simili ad ictus in pazienti di età inferiore ai 40 anni in assenza di fattori di rischio per ictus.
Quadro clinico
MELAS di solito ha un decorso recidivante-remittente, con o senza deficit crescenti. Le manifestazioni cliniche sono caratterizzate da:
- manifestazioni comuni
- episodi simili ad ictus
- convulsioni
- acidosi lattica
- encefalopatia
- demenza
- debolezza muscolare
- sordità
Patologia
Un difetto sotto forma di mutazione puntiforme nel nucleotide 3243 del DNA mitocondriale (traslocazione A - G), che codifica per il tRNA della leucina, colpisce la catena respiratoria (responsabile della produzione di energia). La sintesi di una proteina difettosa porta a interruzioni nel funzionamento di varie parti della catena respiratoria, che alla fine portano all’esaurimento di NAD+ e NADH+. Ciò, a sua volta, porta ad un passaggio metabolico alla glicolisi anaerobica, causando un accumulo di lattato, che rende la corteccia più suscettibile all’ipossia e alla fine porta alla morte neuronale. Poiché alcuni mitocondri vengono trasmessi all'uovo, solo una parte dei mitocondri contiene il DNA mutante. La gravità delle manifestazioni cliniche dipende dal numero di geni mutanti.
Diagnostica
Tomografia computerizzata
- attacchi cardiaci multipli
- calcificazione dei gangli della base
- più pronunciato nei pazienti anziani
- atrofia
Risonanza magnetica
- attacchi cardiaci cronici
- danno a diversi letti vascolari
- può essere simmetrico o asimmetrico
- Le lesioni parieto-occipitale e temporo-parietale sono più comuni
- attacchi cardiaci acuti
- rigonfiamento delle circonvoluzioni con aumento del segnale RM sulle immagini pesate in T2
- possibile l'aumento del contrasto
- lesioni della sostanza bianca sottocorticale
- aumento del segnale sulle immagini pesate in diffusione (transilluminazione T2) con cambiamenti minori (se presenti) sull'IDC: i cambiamenti nel MELAS sono caratterizzati da edema vasogenico piuttosto che citotossico, come nell'infarto
Spettroscopia MR: è possibile aumentare il picco del lattato anche nel parenchima cerebrale e nel liquido cerebrospinale invariati.
Diagnosi differenziale
- altre malattie mitocondriali
- Epilessia mioclonica con fibre muscolari strappate
- Sindrome di Kearns-Sayre
- stato epilettico
- encefalite virale
- vasculite cerebrale
- Malattia di Creutzfeldt-Jakob
- attacco cardiaco dovuto
- embolia
- dissezione
- TsADASIL: focolai non sottocorticali
Punti chiave
La diagnosi di MELAS si basa su quanto segue: 1) episodi simili a ictus che si verificano prima dei 40 anni, 2) encefalopatia con epilessia e/o demenza, 3) presenza di acidosi lattica, danno alle fibre muscolari rosse, nonché criteri aggiuntivi come mal di testa ricorrenti, dolore e vomito ricorrente. I tipici risultati dell'imaging includono focolai simili a ictus, calcificazioni dei gangli della base e atrofia. Posizione del danno non coincidente con i confini dei territori vascolari arteriosi, così come l'età e la mancanza di fattori di rischio, sono contrari all'ictus. Nella sindrome MELAS, il danno è spesso dovuto all'edema vasogenico e l'IDC di solito non è ridotto o è meno ridotto rispetto a un recente infarto.