Колко живеят децата със синдром на Мелас? Синдром MELAS - лечение в Италия
Синдромът на MELAS е митохондриално разстройство, характеризиращо се със засягане на мускулите и ЦНС.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - „митохондриална енцефаломиопатия, лактатна ацидоза, stroke-like епизоди“) е прогресивно невродегенеративно заболяване, характеризиращо се с проявите, изброени в заглавието, и е придружено от полиморфни симптоми - инсулт, диабет, гърчове, намалена загуба на слуха, сърдечни заболявания, нисък ръст, ендокринопатии, непоносимост към упражнения и невропсихиатрични разстройства.
История.
Синдромът MELAS е описан за първи път през 1984 г. от Павлакис и колеги; десет години по-късно Павлакис и Мицио Хирано публикуват преглед на 110 случая.
тип наследяване:
майчина
Епидемиология:
Точната честота на заболяването не е известна. В литературата има малко данни за честотата на заболяването. В Северна Финландия процентът на мутация A3243G е 16,3:100 000.
Патогенеза:
Мутациите на митохондриалната ДНК, които контролират дихателната верига на митохондриите, са придружени от нарушаване на процесите на окислително фосфорилиране, най-важният източник на енергия за метаболитните процеси в клетката.
Клинични проявления
На възраст над 40 години пациентите с MELAS се приемат с клиника на преходна исхемична атака, както и с епилепсия, многократно повръщане, главоболие и мускулна слабост. Тези пациенти често са клинично диагностицирани с деменция.
Младата възраст и липсата на рискови фактори, специфични за инсулт, правят MELAS по-внимателен.
Лабораторни данни
Лактатна ацидоза - повишени нива на лактат и пируват.
Данни за визуализация
Промените в мозъка са подобни на промените при инсулт.
Разлики от инсулт
1) засегнатите области не съвпадат с границите на артериалните съдови басейни.
2) при повтарящи се атаки огнищата се визуализират в различна локализация.
+ клинични данни (млада възраст, липса на рискови фактори за инсулт).
CT
Множество хиподенсни области, несъвместими със съдовото легло.
Калцификация на базалните ганглии (най-често при по-възрастни пациенти).
Атрофията възниква на фона на регресия и клинично подобрение.
ЯМР
Остър инфаркт
За разграничаване с инсулт се използват ADC и DWI (ограничаване на дифузията при инсулти (цитотоксичен оток), а при MELAS дифузията е леко ограничена или непроменена (вазогенен оток).
Участие в патологичния процес на субкортикалното бяло вещество на мозъка.
Влошаване на визуализацията на яснотата на контурите на извивките и увеличаване на сигнала от тях върху Т2-претеглени изображения.
Хроничен инфаркт
Промените могат да бъдат симетрични или асиметрични.
Фокалната атрофия възниква на фона на регресия и клинично подобрение.
Най-често се засягат теменните, тилните и темпоралните дялове на мозъка.
MR спектроскопия
Повишени нива на лактат.






Ключови думи
СИНДРОМ НА МЕЛАС / СИНДРОМ НА МЕЛАС / ЕПИЛЕПСИЯ / ЕПИЛЕПСИЯ / КЛИНИКАанотация научна статия по клинична медицина, автор на научна работа - Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А.
Синдромът MELAS е генетично обусловено заболяване от групата на митохондриалните заболявания, дефинирано като митохондриална енцефаломиопатия с лактатна ацидоза и инсултоподобни епизоди (митохондриална енцефаломиопатия, лактатна ацидоза с инсултоподобни епизоди). Всички органи и тъкани са включени в патологичния процес, но в по-голяма степен страдат мускулната и нервната система. Най-често заболяването се развива във възрастта между 6 и 10 години. Протичането на заболяването е прогресивно. В повечето случаи заболяването се проявява с епилептични припадъци, повтарящи се главоболия, повръщане и анорексия. Епилепсията е важна клинична проява на синдрома MELAS. Епилептичните припадъци са първият разпознаваем симптом при митохондриални енцефалопатии (МЕ) в 53% от случаите. При MELAS най-честа е тилната епилепсия. С прогресирането на заболяването се отбелязва резистентност на епилепсията към терапията, често със статусен курс. Описани са случаи на трансформация в епилепсия на Кожевников. Представяме история на пациент с диагноза MELAS синдром, потвърдена през живота му.
Свързани теми научни трудове по клинична медицина, автор на научна работа - Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А.
-
Митохондриална енцефалопатия с инсулт-подобни епизоди и лактатна ацидоза (мелас синдром): диагностични критерии, характеристики на епилептичните припадъци и подходи за лечение на примера на клиничен случай
2017 / Ямин М.А., Черникова И.В., Арасланова Л.В., Шевкун П.А. -
Инсулти при митохондриални заболявания
2012 г. / Пизова Н.В. -
Епилепсия при деца с митохондриални заболявания: характеристики на диагностика и лечение
2012 / Заваденко Н. Н., Холин А. А. -
Неврологични нарушения при митохондриална енцефаломиопатия - лактатна ацидоза с удароподобни епизоди (MELAS синдром)
2012 / Харламов Дмитрий Алексеевич, Крапивкин Алексей Игоревич, Сухоруков Владимир Сергеевич, Куфтина Людмила Андреевна, Грознова Олга Сергеевна -
Синдромът на Melas като необичайна причина за хипопаратироидизъм: клиничен случай
2018 / Умярова Диляра Шамилевна, Гребенникова Татяна Алексеевна, Зенкова Татяна Станиславовна, Соркина Екатерина Леонидовна, Белая Жана Евгениевна -
Инсулт-подобни епизоди при митохондриална енцефаломиопатия с лактатна ацидоза
2010 / Калашникова Людмила Андреевна, Добринина Л. А., Сахарова А. В., Чайковская Р. П., Мир-касимов М. Ф., Коновалов Р. Н., Шабалина А. А., Костырева М. В., Гнездицки В. В., Процки С. В. -
Митохондриални цитопатии: melas и MIDD синдроми. Един генетичен дефект, различни клинични фенотипове
2017 / Муранова А.В., Строков И.А. -
Доброкачествена тилна епилепсия в детска възраст с ранно начало (синдром на Панайотопулос). Описание на клиничния случай
2015 / Матюк Ю.В., Котов А.С., Борисова М.Н., Пантелеева М.В., Шаталин А.В. -
Полиморфизъм на клиничните прояви на прогресивна митохондриална енцефаломиопатия, свързана с мутация на ген POLG1
2016 / Yablonskaya M.I., Николаева E.A., Shatalov P.A., Kharabadze M.N. -
Диагностична стойност на изследването на цитохимичната активност на ензимите при наследствени митохондриални заболявания
2017 / Казанцева И.А., Котов С.В., Бородатая Е.В., Сидорова О.П., Котов А.С.
ЕПИЛЕПСИЯ ПРИ МЕЛАС СИНДРОМ
Синдромът MELAS е генетично обусловено заболяване от групата на митохондриите, дефинирано като митохондриална енцефаломиопатия, лактатна ацидоза с епизоди, подобни на инсулт. Патологичният процес обхваща всички органи и тъкани, но е неблагоприятен най-вече за мускулната и нервната система. Заболяването е най-често при деца от 6 до 10 години. Клиничното протичане е прогресивно. В повечето случаи заболяването се проявява с епилептични припадъци, рецидивиращи главоболия, повръщане, анорексия. Важното клинично представяне на синдрома на MELAS е епилепсията. Епилептичните припадъци са първоначалният диагностичен симптом на митохондриалните енцефалопатии (МЕ) в 53% от случаите. Окципиталната епилепсия е най-честата при синдрома на MELAS. С напредването на заболяването се наблюдава резистентност на епилепсията към лечението, често с появата на епилептичен статус. Описани са някои случаи на трансформация в епилепсия на Кожевников Дадена е история на пациент с потвърдена приживе диагноза синдром на MELAS.
Текстът на научната работа на тема "Епилепсия при мелас синдром"
ТОМ IV БРОЙ 3 2009г
ЕПИЛЕПСИЯ С МЕЛАС СИНДРОМ
К.Ю. Мухин1, М.Б. Миронов1, Н.В. Никифорова1, Ц.Б. Михайлова2, VA. Чадаев1, АА. Алиханов1-2, Б.Н. Рижков1, А.С. Петрухин1
ЕПИЛЕПСИЯ ПРИ МЕЛАС СИНДРОМ
КЮ. Мухин1, М.Б. Миронов1, Н.В. Никифорова1, С.В. Михайлова2, UA. Чадаев1, АА. Алиханов1-2, Б.Н. Ryzkov1 AS. Петрухин1
1 - Катедра по неврология и неврохирургия, Факултет по педиатрия, Държавна образователна институция за висше професионално образование, Руски държавен медицински университет на Росздрав
2 - руски детски клинична болница
Синдромът MELAS е генетично обусловено заболяване от групата на митохондриалните заболявания, дефинирано като митохондриална енцефаломиопатия с лактатна ацидоза и инсултоподобни епизоди (митохондриална енцефаломиопатия, млечнокисела с инсултоподобни епизоди). Всички органи и тъкани са включени в патологичния процес, но в по-голяма степен страдат мускулната и нервната система. Най-често заболяването се развива във възрастта между 6 и 10 години. Протичането на заболяването е прогресивно. В повечето случаи заболяването се проявява с епилептични припадъци, повтарящи се главоболия, повръщане и анорексия. Епилепсията е важна клинична проява на синдрома на MELA. Епилептичните припадъци са първият разпознаваем симптом при митохондриални енцефалопатии (МЕ) в 53% от случаите. При MELAS най-честа е тилната епилепсия. С прогресирането на заболяването се отбелязва резистентност на епилепсията към терапията, често със статусен курс. Описани са случаи на трансформация в епилепсия на Кожевников. Представяме история на пациент с диагноза MELAS синдром, потвърдена през живота му.
Ключови думи: MELAS синдром, епилепсия, клиника, диагностика, лечение.
Синдромът MELAS е генетично обусловено заболяване от групата на митохондриите, дефинирано като митохондриална енцефаломиопатия, лактатна ацидоза с епизоди, подобни на инсулт. Патологичният процес обхваща всички органи и тъкани, но е неблагоприятен най-вече за мускулната и нервната система. Заболяването е най-често при деца от 6 до 10 години. Клиничното протичане е прогресивно. В повечето случаи заболяването се проявява с епилептични припадъци, рецидивиращи главоболия, повръщане, анорексия. Важното клинично представяне на синдрома на MELAS е епилепсията. Епилептичните припадъци са първоначалният диагностичен симптом на митохондриалните енцефалопатии (МЕ) в 53% от случаите. Окципиталната епилепсия е най-честата при синдрома на MELAS. С напредването на заболяването се наблюдава резистентност на епилепсията към лечението, често с появата на епилептичен статус. Описани са някои случаи на трансформация в епилепсия на Кожевников Дадена е история на пациент с потвърдена приживе диагноза синдром на MELAS.
Ключови думи: MELAS синдром, епилепсия, клинична картина, диагностика, лечение.
Синдромът MELAS е генетично обусловено заболяване от групата на митохондриалните заболявания, дефинирано като митохондриална енцефаломиопатия с лактатна ацидоза и инсултоподобни епизоди (митохондриална енцефаломиопатия, лактатна ацидоза с инсултоподобни епизоди).
Синдромът MELAS е идентифициран за първи път като независима нозологична форма от S. Pavlakis et al. през 1984 г. Въпреки това, редица автори предполагат, че заболяването е описано по-рано под името "фамилна полиодистрофия, митохондриална миопатия, млечна ацидемия".
Разпространението в популацията не е установено. До 2000 г. бяха публикувани повече от 120 наблюдения на синдрома на MELAS, включително в местната преса.
Синдромът MELAS в 25% от случаите се наследява по майчина линия с висок риск, но при 56-75% от пациентите фамилната анамнеза не е обременена. Заболяването е свързано с мутации в гени на митохондриална ДНК, кодиращи субединици на комплекси на дихателната верига и гени на транспортна РНК (MT-ND1, MT-ND5, MT-TH, MT-TL1 и MT-TV). В 80-90% от случаите на синдрома на MELAS заболяването се основава на точкова мутация в гена MT-TL1, кодиращ левцинова трансферна РНК. При тази мутация адениновият нуклеотид се заменя с гуанин на позиция 3243 (A3243G), което нарушава синтеза на всички протеини в митохондриите.
Всички органи и тъкани са включени в патологичния процес, но в по-голяма степен страдат мускулната и нервната система.
Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова Ц.В., Чадаев В.А., Алиханов А.А., Рижков Б.Н., Петрухин А.С.
Епилепсия при синдром на MELAS Rus. жур. дет. Невр.: т. IV, бр. 3, 2009 г.
ОРИГИНАЛНИ СТАТИИ
теми като най-нестабилни. тежест клинични проявлениязависи от праговия ефект (възраст, енергийни нужди на тъканите), от контрола на ядрените гени върху синтеза на дихателната верига, хетероплазмия (различно съдържание на мутантни mtDNA молекули в тъканите). Доказано е, че при пациенти със синдрома MELAS съдържанието на мутантна mtDNA в различни тъкани е 93-96%. В членовете на семейството на пробанд също се открива мутантна mtDNA в тъканите, но съдържанието му е значително по-ниско: 62-89% в изтритата форма на заболяването, от 28 до 89% при липса на клинични признаци на синдрома.
Най-често заболяването се развива на възраст от 6 до 10 години, но има случаи на по-ранен (до две години) или по-късен дебют - от 21 до 40 години. Преди началото на заболяването 90-100% от пациентите се развиват нормално. Протичането на заболяването е прогресивно, по-злокачествено с ранно начало.
В повечето случаи заболяването се проявява с епилептични припадъци, повтарящи се главоболия, повръщане и анорексия. Трябва също да обърнете внимание на непоносимостта към физическа активност под формата на влошаване на здравето и появата на мускулна слабост. Комплексът от миопатични симптоми се проявява чрез непоносимост към физическо натоварване, мускулна слабост, умора и понякога мускулна хипотрофия.
С напредването на заболяването обикновено се развива деменция. По-рядко се срещат симптоми като церебеларна атаксия, невросензорна глухота и периферна полиневропатия.
Характерни са епизоди, подобни на инсулт, които могат да се проявят чрез повтарящи се пристъпи на главоболие, замаяност, развитие на фокални неврологични симптоми (пареза, хемианопсия) и кома. Тези остри епизоди често се предизвикват от треска или интеркурентни инфекции. Тези прояви могат да имат доста бърза регресия (от няколко часа до няколко седмици), както и тенденция към рецидив.
Епилепсията е важна клинична проява, която често се появява в ранните стадии на MELAS. то
често най-очевидната неврологична проява, особено при атипична митохондриална енцефалопатия (МЕ). Епилептичните припадъци са първият разпознаваем симптом при митохондриални енцефалопатии (МЕ) в 53% от случаите.
При MELAS най-честата е окципиталната епилепсия (SE). Характеризира се с фокални гърчове, произхождащи от тилните дялове. Припадъците често са свързани с преходни или персистиращи неврологични симптоми като загуба на зрително поле.
Припадъците, произтичащи от тилната кора, се разделят според техните прояви на субективни усещания (аура) и клинично откриваеми симптоми, като правило, с двигателен компонент. Епилептичните аури, излъчвани от тилния лоб, включват прости и сложни зрителни халюцинации, амавроза. Най-типичните гърчове, характерни за SE, са прости зрителни халюцинации, които могат да се проявят като положителни (светкавици, светлинни петна) и отрицателни симптоми (скотома, хемианопсия). Най-често зрителните халюцинации се описват като петно или петна светлина, постоянни или мигащи. По правило петното е бяло със зеленикав оттенък. Също така халюцинациите могат да бъдат многоцветни или монохроматични. Халюцинациите обикновено се появяват в зрителните полета контралатерално на фокуса на възбуждане в тилната кора с последващо разпространение. Все пак трябва да се отбележи, че в оплакванията на пациентите визуалната аура не се открива често.
Сложни зрителни халюцинации се наблюдават, когато епилептичното възбуждане се разпространява в окципито-темпоралната или окципито-париеталната област. Комплексните зрителни халюцинации могат да се появят под формата на хора, животински обекти или сцени, да бъдат познати или непознати, приятни или ужасяващи, плашещи, прости или гротескни, могат да бъдат статични или да се движат в хоризонтална равнина и да изчезнат. По правило те са терминален симптом преди развитието на моторна атака; може да бъде първият иктален симптом, но по-често се появява след него
ТОМ IV БРОЙ 3 2009г
основни халюцинации.
Ictal ama vrosis е особен, изключително труден за диагностициране тип припадъци, излъчвани от тилната кора. Според много автори това е същият често срещан симптом на дразнене на тилната част, както и зрителни халюцинации, но често остава неразпознат. Обикновено пациентите не разграничават този симптом отделно в структурата на атаката. Загубата на зрение възниква двустранно със загуба на страничните полета. Възможна хомонимна хемианопия, контралатерална на фокуса на атаката. Усещанията на пациентите се описват от тях като притъмняване в очите, "бял мрак", нарушено цветоусещане. Може би статусен курс с образуването на така наречения епилептичен статус amauroticus.
Окципиталните гърчове могат да се проявят с автономни симптоми. Те включват мигренозно главоболие, виене на свят, гадене и повръщане. Често срещан симптом е главоболие, подобно на мигрена след пристъп.
Клиничните прояви на гърчове, които се появяват ограничено в тилната кора, се характеризират с отклонение на очите встрани. Отклонението на очите може да се отбележи заедно с отклонението на главата настрани. В повечето случаи се отбелязва отклонение на очите към контралатералния фокус. Описани са обаче случаи, когато се наблюдава отвличане на очите към фокуса. Също така, една от характеристиките на "тилните" припадъци е мигновеното разпределение на изхвърлянето към предните части на мозъка, докато клиничната картина, като правило, е доминирана от изразен двигателен компонент. Възможни са тонични, тонично-клонични (както хемиконвулсивни, така и вторично генерализирани), автомоторни гърчове. В тази връзка е важно да се идентифицират първоначалните клинични симптоми - немотивирано и внезапно спиране на погледа, гледане на несъществуващи обекти, неразумна усмивка, вегетативни прояви и задължително документиране на първичната иктогенна зона с помощта на метода VEM.
С прогресирането на заболяването се отбелязва резистентност на епилепсията към терапията, често със статусен курс. Описани са случаи на трансформация в епилепсия на Кожевников. Редица авто-
Rov описва възможността за епилептичен статус като първи симптом при пациенти с MELAS без анамнеза за предишни епилептични припадъци. Ribacoba R. et al. описват в публикацията си 4 случая на развитие на epilepsia partialis continua с фокални моторни припадъци, предшествани от анамнеза за епизоди на мигренозно главоболие. Miyazaki M. и др. показаха възможността за продължаващ фокален миоклонус в рамките на epilepsia partialis continua при пациенти с MELAS. Араки Т. и др. наблюдава пациент на възраст 37 години с епилептичен статус на фокални припадъци под формата на колебания на съзнанието, хомонимна хемианопсия в комбинация с пароксизмални епизоди на отклонение на окото встрани. ЕЕГ регистрира продължителни ЕЕГ модели на гърчове, локализирани в тилната област. При възрастни пациенти с MELAS има преобладаване на фокални моторни припадъци, но ЕЕГ показва преобладаване на мултирегионална епилептиформена активност в тилната област.
Епилептиформната активност се регистрира в 71% от случаите след началото на пристъпите. Електроенцефалографско изследване на пациенти със синдром на MELAS се характеризира с епилептиформна активност в тилната област. Редица автори свързват появата на регионални епилептиформни разстройства с инсулти. Според изследването на Fujimoto S., в острия период (т.е. в рамките на 5 дни след инсулт-подобен епизод) по-голямата част от изследваните пациенти със синдром на MELAS са имали регионални делта вълни с висока амплитуда в комбинация с полипикове. Авторите предлагат този модел да се разглежда като патогномоничен за подобни на инсулт епизоди. В допълнение към тилната област, епилептиформната активност може да се разпространи в темпоралните области, бифронтално, а също и двустранно в задните области с дифузно разпространение. Може би появата на фотопароксизмен отговор по време на ритмична фотостимулация.
Водещият лабораторен признак е повишаване на нивото на лактат в кръвта.
ОРИГИНАЛНИ СТАТИИ
wi над 2,0 mmol/l, което води до развитие на лактатна ацидоза.
MRI на мозъка в ранните стадии на заболяването може да бъде незабележимо, дори ако се появи епилепсия. Невроизобразителните методи разкриват инфарктни зони в мозъчните хемисфери (80%), по-рядко в малкия мозък и базалните ганглии. Може да има и калцификация на базалните ганглии, атрофия на кората на главния мозък. При изследване на фотонна емисия натрупването на изотопа се открива 3-16 дни преди появата на инфарктната зона (намаляване на изотопния сигнал) на компютърна томограма на мозъка. ЯМР на мозъка показва лезии, локализирани предимно в тилните лобове, които могат да бъдат преходни. Засяга се предимно тилната кора, в по-малка степен се уврежда бялото вещество. На Т2-претеглени изображения мозъчните лезии при MELA се появяват като области с повишен интензитет на сигнала. Преходните хиперинтензивни зони се свързват от редица автори с обратим съдов оток.
Ангиографията обикновено не разкрива съдови аномалии. Дифузионно претегленият ЯМР показва промени, свързани с вазогенен оток.
Хистопатология: Мускулна биопсия разкрива влакна с разкъсани "червени ръбове". Аутопсията на мозъка се характеризира с комбинация от стари и нови огнища на инфаркти, както и атрофия на кората с фокални огнища на некроза.
В момента терапията е поддържаща. Основната посока на лечението е подобряване на енергийния баланс на митохондриите и дихателната верига. Прилагайте коензим p10 (80-300 mg / ден), витамини K1 и KZ (25 mg / ден), янтарна киселина (до 6 g / ден), витамин С (2-4 g / ден), рибофлавин (100 mg / ден). ден) и никотинамид (до 1 g/ден). Във връзка с развиващия се вторичен дефицит на карнитин, на пациентите се предписва L-карнитин (до 100 mg / kg / ден). Като антиоксидантна терапия се използват витамин Е (300-500 mg/ден) и витамин С (2-4 mg/ден).
Няма общоприети антиепилептични терапевтични схеми за MELA. Редица автори предлагат да се изключат лекарства, които могат да инхибират енергийния метаболизъм (барбитурати, лекарства с валпроева киселина; както и някои лекарства от други групи, например хлорамфеникол). В литературата са описани няколко изолирани случая на влошаване на гърчове с употребата на валпроева киселина при синдрома MELA с мутация A3243C. Основните AED при лечението на епилепсия при синдрома на MELA са тегретол (или трилептал), топамакс, кепра в средни терапевтични дози. Правилно избраната терапия води до значително намаляване на честотата на вторичните генерализирани конвулсивни припадъци. Въпреки това, гърчовете с нарушени вегетативно-висцерални и зрителни функции обикновено са резистентни на лечение. В терминалния стадий на заболяването честотата на епилептичните припадъци може да намалее.
Ето историята на случай на пациент с диагноза синдром на MELAY, потвърдена през живота му.
Пациент Ч.А., на 11 години, е наблюдаван в Центъра по детска неврология и епилепсия. При приемане се оплакват от постепенна загуба на говорни умения, изразено нарушение на походката с отказ от ходене, значително намаляване на зрението, капризност и негативно поведение; ежедневни серийни атаки под формата на потрепване на мускулите на лицето, мускулите на горните и долните крайници, както и краткотрайни епизоди на загуба на зрение.
Дебютът на заболяването е отбелязан на възраст 5 години и 9 месеца. За първи път, на фона на пълно здраве, при заспиване се появи силно главоболие, прости зрителни халюцинации ("жълт лъч"), последвани от бурно завъртане на очите и главата настрани и развитие на генерализиран тонично-клоничен конвулсивен припадък, след което се забелязва повръщане. След 9 месеца атаките със същите симптоми се повтарят и бързо придобиват сериен характер. След назначаването на тегретол в доза от 400 mg на ден, честотата на атаките намалява до 1 път на месец. Tegretol е заменен с Depakine Chrono в доза 900 mg/ден, на фона на който е отбелязана клинична ремисия за 6 месеца. Като се има предвид клиничният симптом
ТОМ IV БРОЙ 3 2009г
томатика, ограничаване на припадъците до периода на заспиване, нормален интелект на пациента, положителна реакция към валпроат, диагностицирана е идиопатична окципитална епилепсия.
На 7-годишна възраст фокалните версивни пристъпи се възобновяват с вторична генерализация при заспиване със същата честота 1 път на месец. Увеличаването на дозата на Depakine до 1500 mg/ден не води до намаляване на честотата на гърчовете. При добавяне на lamiktal в доза от 75 mg/ден пристъпите спират за 4 месеца, след което се възобновяват със същата честота. На 8-годишна възраст се присъединяват пристъпи с краткотрайна загуба на зрение. От 8 години 8 месеца преди заспиване започнаха да се появяват нетипични абсанси: бързо мигане със затваряне на клепачите и издигане на очните ябълки нагоре; съзнанието се колебае.
На 9-годишна възраст се появяват множество серийни атаки, продължаващи няколко дни, с прости зрителни халюцинации под формата на мигащ "лъч" пред очите, с обръщане на очите и главата надясно. Преди заспиване такива атаки понякога се превръщат във фокални хемиклонични, които се проявяват чрез намаляване на лицевата
мускулатура вдясно, потрепване на главата вдясно, клонии на десните крайници (по-големи от ръката). Понякога след атаката имаше силно главоболие и повръщане. На същата възраст се появяват инхибиторни припадъци: аура под формата на настръхване палецдесен крак, последван от моментна слабост на десния крак и тромавост на дясната ръка. Топамакс е въведен в схемата на лечение в доза 100 mg / ден - няма епилептични припадъци за 1 година.
Също така на 9-годишна възраст за първи път се появяват пароксизмални състояния, придружени от силно главоболие, повръщане и развитие на дясностранна хемипареза. В някои случаи такива състояния са придружени от амавроза с продължителност от няколко минути до няколко дни.
На 10,5 години атаките се появяват отново под формата на завъртане на главата наляво, резки движения на очните ябълки наляво, с продължителност до 5 s, честота до 3 пъти на час, ежедневно, дори по време на сън. Дозата на Topamax е увеличена до 150 mg/ден без значим ефект. На 10 години и 10 месеца. след интензивно главоболие, редуващи се
Ориз. 1. Пациент Ch.A. 10 години. Диагноза: синдром на MEAE. Симптоматична фокална епилепсия.
Видео-ЕЕГ мониториране (2004): на фона на дифузно забавяне на основната мозъчна дейност се записва продължаваща епилептиформна активност в лявата тилна област. Регистрирани са и субклинични ЕЕГ модели на пристъп в лявата тилна област с разпространение в лявата задна темпорална област.
Център по детска неврология и епилепсия
под ръководството на професор К.Ю. Мухина се занимава с диагностика и лечение на тревожни разстройства на нервната система в Aetei, специализирана в Aetian форми на епилепсия.
Основни направления
дейности:
Епилепсия при деца и юноши
Нарушения на съня при деца
Тики, енуреза
Преглед на деца през първите ^ месеца от живота.
Прегледи в нашия център:
Диагностика и лечение на заболявания на нервната система при деца
Пълна диагностика (включително предоперативна) и лечение на епилепсия
Консултация с невролози и епилептолози
Консултация с педиатър (често боледуващи деца, гастроентеролог и др.)
Консултация с психиатър и психолог.
Генетична консултация с тестове (включително кариотипиране)
Видео-ЕЕГ мониториране (в специално оборудвани стаи на Центъра или с посещение в дома на пациента)
Компютърна (цифрова) електроенцефалография
UZDG (ултразвукова доплерография) на съдовете на главата и шията
Ехоенцефалография (ECHO EG)
На нашия сайт можете да се абонирате за списанието "Руски журнал за детска неврология" чрез Интернет.
Подробна информация за работата на Центъра от 10:00 до 19:00 часа на телефон:
Тел.: (+7495) 983-09-03; (+7926)290-50-30 Тел./Факс: (+7495) 394-82-52
Адрес: ул. Борисовские пруди, 13, бл. 2. Интернет: www.epileptologist.ru Електронна поща: [имейл защитен](за подробна карта на маршрута вижте уебсайта)
ТОМ IV БРОЙ 3 2009г
фокални хемклонични и вторично генерализирани гърчове, които стават серийни и продължават 48 часа. Към топамакс беше добавен Frizium в доза 10 mg/ден с временен положителен ефект.
От 8-годишна възраст започват да се забелязват трудности при усвояването на училищния материал; намалена памет. Имаше повишена умора, изтощение, инхибиране на умствената дейност. Момчето стана капризно, раздразнително, негативно; фонът на настроението е намалял. От 9-годишна възраст се наблюдава засилване на тази симптоматика.
От житейската анамнеза е известно, че детето е родено от втора нормална бременност, раждане на втори термин, тегло при раждане 2800 гр., дължина 53 см. Ранното психомоторно и речево развитие е напълно съобразено с възрастта. Минали заболявания: варицела на 6 години, чести остри респираторни вирусни инфекции (до 4 пъти годишно) от 6 години. Наследствеността за епилепсия и други неврологични заболявания не е обременена.
Към момента на прегледа (11 г.) състоянието на детето е тежко; реагира отрицателно на проверката. Осъзнат, проориентиран
пространство и време. Той влиза в контакт изключително неохотно, отказва да следва инструкциите. Спонтанен нистагъм наляво, глава наклонена към лявото рамо със завъртане надясно. Езикът е в средната линия, фарингеалният рефлекс е намален; Отбелязват се дисфагия и дизартрия. Зрението е намалено.
Определя се умерена дифузна мускулна хипотония. Сухожилните рефлекси са равномерно намалени. Имаше леко намаляване на мускулната сила в десните крайници. Патологични рефлекси на краката не са открити. Няма обективни данни за нарушение на чувствителността. Не си струва в теста на Ромберг. Отказва да ходи. Когато се опитате да го изправите на крака, той плаче, сяда на пода. Липсва при извършване на тест с показалец. Говори бавно, с отделни думи, неохотно.
Допълнителни методипрегледи. Видео-ЕЕГ мониториране (2004). Значително забавяне на основната фонова записваща дейност. По време на изследването е регистрирана продължителна епилептиформна активност в лявата тилна област с разпространение към лявата задна темпорална област и с периодично образуване на ЕЕГ модел
роден 1993г 16/12/05
Ориз. 2. Пациент Ч.А. 11 години. Диагноза: синдром на MELAS. Симптоматична фокална епилепсия.
Видео-ЕЕГ наблюдението се извършва в динамика след 1 година (2005 г.): значително забавяне на фоновата активност на мозъка. По време на записа на съня се регистрира продължително регионално забавяне в дясната фронто-централна област, в структурата на която се открива пикова вълнова активност в дясната фронто-централна област.
ОРИГИНАЛНИ СТАТИИ
ступа (фиг. 1). Също така се определя продължително регионално забавяне в дясната фронто-централна област с включване на единични остри вълни.
Видео-ЕЕГ мониториране в динамика (2005): Значително забавяне на фоновата активност на мозъка. Проучването регистрира продължително регионално забавяне в десния фронто-централен регион. В структурата на регионалното забавяне в дясната фронто-централна област се разкрива пиково-вълнова активност (фиг. 2).
ЯМР на мозъка. Първият ЯМР (6 години) разкрива единичен хиперинтензивен сигнал в Т2 режим в лявото полукълбо на малкия мозък. Проучване с ЯМР във времето (10,5 години): разкрито е значително влошаване на първичната лезия с широко разпространение на патологичния процес в лявата и дясната тилно-париетална област на двете полукълба на мозъка (проф. А. А. Алиханов).
Зрителни евокирани потенциали: значителни морфологични и функционални промени в зрителната аферентна система на ниво зрителен нерв и кортикална част на зрителния анализатор, по-изразени вляво.
Консултация с офталмолог: частична атрофия на зрителните нерви. Елементи на кортикална агнозия.
Електрокардиограма: ектопичен ритъм с ускорение до 100 удара в минута.
Вертикално положение на електрическата ос на сърцето. Промени в процесите на реполяризация, които са по-изразени при ортостаза.
Електроневромиография: разкрива първичния мускулен тип лезия. Скоростите на провеждане по периферните нерви не са намалени.
Изследване на нивото на лактат в кръвта: съдържанието на лактат в кръвта е 3,0 mmol / l (нормата е до 1,8).
Като се има предвид наличието на епилептични припадъци, произтичащи от тилната област на мозъчната кора, резистентни на терапия, инсулт-подобни епизоди, периоди на амавроза, когнитивен спад, наличието на хиперинтензивни сигнали в малкия мозък и задните области на мозъчната кора на ЯМР, предполага се повишаване на нивото на лактат в кръвта, пациентът е имал диагноза MELAS синдром. По време на генетично изследване в кръвните клетки е открита мутация A3243G в хетероплазмено състояние (диагностиката е извършена в Държавния изследователски център на Руската академия на медицинските науки) и диагнозата е потвърдена.
Наблюдението при проследяване показа бърза прогресия на нарушения на висшите психични функции, развитие на кортикална слепота, пълна неподвижност на пациента, последвано от настъпване на смърт на възраст от 12 години и 10 месеца. (след 7 години от началото на заболяването).
Библиография
1. Николаева Е.А., Темин П.А. Митохондриални заболявания, придружени от нарушено нервно-психическо развитие. Синдром MELAS // Наследствени нарушения на нервно-психическото развитие на децата. Ръководство за лекари, редактирано от Temin P.A. Казанцева Л.З. - Медицина, 2001. - С. 96-107.
2. Николаева Е.А., Темин П.А., Никанорова М.Ю., Клембовски А.И., Сухоруков В.С., Дорофеева М.Ю., Корсунски А.А. Лечение на дете с митохондриален синдром MELAS (митохондриална енцефалопатия, лактатна ацидоза, подобни на инсулт епизоди) // Руски бюлетин по перинатология и педиатрия. - 1997. - № 2. - С. 30-34.
3. Смирнова И.Н., Кистенев Б.А., Кротенкова М.В., Суслина З.А. Инсултоподобен курс на митохондриална енцефаломиопатия (MELAS синдром) // Атмосфера. Нервни заболявания. - 2006. - № 1. - С. 43-48.
4. Темин П.А., Никанорова М.Ю., Николаева Е.А. Синдром на MELAS (митохондриална енцефаломиопатия, лактатна ацидоза, епизоди, подобни на инсулт): основни прояви, диагностични критерии, възможности за лечение // Nevrol. списание - 1998. - № 2. - С. 43-48.
5. Ajmone-Marsan C., Ralston B. Епилептичният припадък, неговата функционална морфология и диагностично значение. - Спрингфийлд (Илинойс): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Иктална кортикална слепота с постоянна загуба на зрение // Епилепсия. - 1989. - Т. 30. - С. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Случай на MELAS, представящ сложен частичен епилептичен статус // Rinsho Shinkeigaku. - 2001. - V. 41 (8). - С. 487-90.
ТОМ IV БРОЙ 3 2009г
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic фенотипове, свързани с митохондриални нарушения // Неврология. - 2001. - V. 56 (10). - С. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Валпроевата киселина влошава епилепсията, дължаща се на MELAS при пациент с A3243G мутация на митохондриална ДНК // Metab Brain Dis. - 2007 - V. 22(1). - С. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Молекулярна патология на MELAS и MERRF. Връзката между мутационния товар и клиничните фенотипове // Мозък. - 1997. - Т.120. - С. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Полиморфизъм на епилепсия, свързан с A3243G мутация на митохондриална ДНК (MELAS): причини за забавена диагноза // Rev Neurol (Париж). - 2004. - V. 160 (8-9). - С. 824-829.
12. Дворкин Г., Андерман Ф., Карпентър С. Класическа мигрена, трудноразрешима епилепсия и множествени инсулти: синдром, свързан с митохондриална енцефалопатия / В: Андерман Ф., Лугареси Е., редактори. мигрена и епилепсия. - Бостън: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Серийни електроенцефалографски находки при пациенти с MELAS // Pediatr Neurol. - 1999. - V. 20(1). - С. 43-48.
14. Goto Y., Nonaka I., Horai S.A. Мутация в гена tRNA leu (UUR), свързана с MELAS подгрупата на митохондриалните енцефаломиопатии // Nature. - 1990. - V. 348. - P. 651-653.
15. Хасуо К., Тамура С., Ясумори К., Учино А., Года С., Ишимото С. и др. Компютърна томография и ангиография при MELAS (митохондриална миопатия, енцефалопатия, лактатна ацидоза и подобни на инсулт епизоди): доклад за 3 случая // Неврорадиология. - 1987.-V. 29. - С. 393-397.
16. Хирано М., Павлакис С.Г. Митохондриална миопатия, енцефалопатия, лактатна ацидоза и епизоди, подобни на инсулт (MELAS): Актуални концепции // J. clin. неврол. - 1994. - Т. 9. - С. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Епилептични припадъци при пациент с митохондриална миопатия, енцефалопатия, млечна киселина и инсулт-подобни епизоди ( MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43 (3). - С. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J. и др. Митохондриална енцефаломиопатия с повишение на лактат-пируват и мозъчни инфаркти // Неврология. - 1984. - Т. 34. - С. 72-77.
19. Kuzniecky R. Симптоматична епилепсия на тилния лоб // Епилепсия. - 1998. - V. 39 Suppl 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Дълбочина и директен кортикален запис при гърчове с екстратемпорален произход // Неврология. - 1976. - Т. 26. - С. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Клинични иктални модели при пациенти с епилепсия с тилни електроенцефалографски фокуси // Неврология. - 1975. - Т. 25. - С. 463-471.
22. Матюс П.М., Тампиери Д., Беркович С.Ф., Андерман Ф., Силвър К., Читят Д. и др. Магнитно-резонансното изображение показва специфични аномалии при синдрома на MELAS // Неврология. - 1991. - Т. 41. - С. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Случай с MELAS, свързан с epilepsia partialis continua // No To Hattatsu. - 1991. - V. 23 (1). - С. 65-70.
24. Монтаня П., Галаси Р., Медори Р., Говони Е., Зевиани М., Ди Мауро С. и др. Синдром на MELAS: характерни мигренозни и епилептични характеристики и предаване от майката // Неврология. - 1988. - Т. 38. - С. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N. и др. Церебрален кръвен поток при митохондриална миопатия, енцефалопатия, лактатна ацидоза и подобни на инсулт епизоди // Инсулт. - 1993. - Т. 24. - С. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Митохондриална миопатия, енцефалопатия, лактатна ацидоза и епизоди, подобни на инсулт: отличителен клиничен синдром // An neurol. - 1984. - Т. 16. - С. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Характеристики на епилептичен статус при MELAS. Анализ на четири случая // Neurologia. - 2006. - V. 21 (1). - С. 1-11.
28. Williamson P.D., Spencer S.S. Клинични и ЕЕГ характеристики на сложни парциални припадъци с екстратемпорален произход // Епилепсия. - 1986. - V. 27 (Suppl 2). - С. 46-63.
29. Уилямсън П.Д., Тадани В.М., Дарси Т.М., Спенсър Д.Д., Спенсър С.С., Матсън Р.Х. Епилепсия на тилния лоб: клинични характеристики, модели на разпространение на припадъци и резултати от операция // Ann Neurol. - 1992. - Т. 31. - С. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Парадоксален ефект на натриев валпроат, който влошава епилепсията на MELAS при пациент с A3243G мутация на митохондриалната ДНК // Central European Journal of Medicine. - 2007. - Т. 2(1). - С.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Вазогенен оток на MELAS: серийно проучване с дифузно-претеглено MR изображение // Неврология. - 1999. - Т. 53. - С. 2182-2184.
MELAS синдром(от английски. Митохондриална енцефаломиопатия, лактатна ацидоза и епизоди, подобни на инсулт) - митохондриална енцефаломиопатия, лактатна ацидоза и подобни на инсулт епизоди - обикновено се проявява след 5-20 години. Заболяването се проявява предимно чрез остри удароподобни епизоди с развитие на фокални промени в тилната и теменно-темпоралната област на мозъка и появата на подходящи неврологични симптоми (пареза, кортикални зрителни нарушения, конвулсии, кома, главоболие и повръщане). и т.н.). Появата на огнища е свързана с преходна дисфункция на окислителното фосфорилиране в мозъчния паренхим, както и структурни и метаболитни нарушения в стените на артериолите и капилярите; характерна особеносттакива "смесени" в genszu мозъчни инфаркти е относително бързо възстановяване.
За синдрома на MELASмиопатични прояви (умора и непоносимост към упражнения), деменция, атаксия, дегенерация на ретината, сензоневрална глухота, нисък ръст, диабет, кардиомиопатия и редица други мултиорганни прояви също могат да бъдат наблюдавани. Характерно е значително ниво на лактатна ацидоза в кръвта и цереброспиналната течност, при биопсия на скелетните мускули често се разкрива феноменът на "разкъсани червени влакна". MELAS се унаследява по майчина линия, но изключителната променливост на клиничните прояви може да затрудни оценката на фамилната история.
При пациенти със синдром на MELASописват най-малко 8 точкови мутации в гените на mtDNA, като 5 от тях са локализирани в различни региони на tRNA гена). Най-честата мутация е A->G заместването на позиция 3243 (около 80% от пациентите) и като цяло мутациите на този левцинов tRNA ген се откриват в почти 95% от случаите на MELAS. В редки случаи при пациенти с MELAS са описани точкови мутации в гените на други тРНК и СОХ гена на III субединица на комплекс IV на респираторната верига. Всички мутации се откриват в хетероплазмено състояние.
NARP синдром(от английски Neuropathy, Ataxia, Retinitis Pigmentosa) - невропатия с атаксия и ретинит пигментоза - характеризира се, в съответствие с името, с развитието на прогресивна периферна невропатия с мускулна слабост, церебеларна атаксия и пигментна дегенерация на ретината. Както при други митохондриални енцефаломиопатии, клиничната картина може да бъде много променлива, с наличие или липса на редица допълнителни симптоми при роднини (забавено психомоторно развитие, епилептични припадъци, деменция). Изследванията за лактатна ацидоза и други маркери на митохондриална дисфункция не винаги са информативни.
Тип наследяване на болестта майчина. Всички пациенти с NARP синдром имат хетероплазмена мутация T=>G на позиция 8993 (ATPase 6 ген - субединица V на комплекса на дихателната верига). Нивото на хетероплазма е определящо за естеството на проявлението на тази мутация: когато съдържанието на мутантната mtDNA<78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея .
 Съдържание на темата "Митохондриална патология на нервната система":
Съдържание на темата "Митохондриална патология на нервната система": Синдромът MELAS се отнася до митохондриални заболявания (MD), които се причиняват от генетични и структурно-биохимични дефекти на митохондриите и са придружени от нарушение на тъканното дишане и в резултат на това системен дефект в енергийния метаболизъм, в резултат на което повечето енергозависими тъкани и целеви органи са засегнати в различни комбинации: мозък, скелетни мускули и миокард, панкреас, орган на зрението, бъбреци, черен дроб. Клинично нарушенията в тези органи могат да се проявят във всяка възраст. В същото време хетерогенността на симптомите усложнява клиничната диагноза на тези заболявания. Необходимостта от изключване на MB възниква при наличие на мултисистемни прояви, които не се вписват в обичайния патологичен процес. Честотата на дисфункция на дихателната верига се оценява от 1 на 5-10 хиляди до 4-5 на 100 хиляди новородени.
прочетете и поста: Митохондриални заболявания(към уебсайта)
Синдромът MELAS (митохондриална енцефаломиопатия, лактатна ацидоза и инсулт-подобни епизоди) е мултисистемно заболяване, характеризиращо се с инсулт-подобни епизоди, които се появяват в млада възраст (до 40 години), енцефалопатия с гърчове и деменция, митохондриална миопатия с феномена " разкъсани" червени влакна и лактатна ацидоза (възможно е повишаване на нивото на млечна киселина в кръвта без ацидоза).
Синдромът на MELAS се основава на точкови мутации в митохондриалната ДНК (mtDNA). Заболяването се наследява по майчина линия (следователно роднините по майчина линия вероятно са носители на такива мутации; по-често роднините по майчина линия описват олигосимптомна клинична картина с отделни симптоми на синдрома на MELAS; при асимптоматични роднини синдромът на MELAS се идентифицира само чрез резултатите от мускулна биопсия или молекулярно изследване). Понастоящем са известни повече от десет гена, чиито мутации водят до развитието на клиничната картина на синдрома MELAS. В повечето случаи развитието на синдрома MELAS се причинява от мутации в гените, кодиращи функциите на трансферната РНК.
Обикновено заболяването дебютира на възраст от 6 до 10 години (възрастта на началото на заболяването е от 3 до 40 години; ранното начало на заболяването е типично и се среща при 90% от пациентите). Характеризира се с нисък растеж на пациентите (и непоносимост към физическа активност). От страна на вътрешните органи могат да се наблюдават кардиомиопатия, нарушена сърдечна проводимост, захарен диабет, нефропатия, нарушена мотилитета на стомашно-чревния тракт.
Помня! Основните клинични критерии за диагностициране на MELAS са: [ 1 ] тип наследство по майчина линия; [ 2 ] започнете преди 40-годишна възраст; [ 3 ] нормално психомоторно развитие преди заболяване; [ 4 ] непоносимост към физическа активност; [ 5 ] мигреноподобно главоболие с гадене и повръщане; [ 6 ] инсулт-подобни епизоди; [ 7 ] енцефалопатия с епилептични припадъци и / или деменция (най-често се регистрират миоклонични припадъци, но се отбелязват и фокални сензорни, моторни и вторично генерализирани тонично-клонични припадъци); [ 8 ] лактатна ацидоза; [ 9 ] разкъсани червени влакна в биопсии на скелетни мускули; [ 10 ] прогресивен курс.
Характерната клинична характеристика на синдрома MELAS са инсулт-подобни епизоди (IPE), които са причина за внезапното развитие на фокални неврологични разстройства. Характерна особеност на IPE е "задната" локализация на лезиите в мозъка. Най-често лезиите се локализират в тилния, париеталния и темпоралния лоб, по-рядко във фронталния лоб, малкия мозък или базалните ганглии; често те са множество. Селективността на локализацията на фокалните промени определя особеностите на фокалните неврологични симптоми: хемианопсия, сензорна афазия, акалкулия, аграфия, оптико-пространствени нарушения, атаксия, промени в съзнанието ([ !!! ] най-често огнищата се локализират в кората на тилните дялове на мозъчните хемисфери, което води до хемианопия или кортикална слепота). Инсултите могат да бъдат разрешени или дългосрочно определени под формата на клинични и / или рентгенологични промени (в зависимост от тежестта на метаболитните нарушения, дължащи се на енергиен дефицит на невроните). Често повтарящи се "мозъчни инфаркти" се развиват с интервал от 1 - 3 месеца в симетрични области. Тези огнища могат да бъдат малки или големи, единични или множествени, обикновено са асиметрични и локализацията им не съответства на зоната на кръвоснабдяване. В допълнение, пациентите със синдром на MELAS могат да имат калцификати в базалните ганглии (в тези случаи CT на мозъка може да осигури диагностична помощ). В неврологичния статус тези морфологични промени се проявяват с миоклонус, атаксия, епизоди на остра психоза или нарушено (дефицитно) съзнание до кома ([ !!! ] характеристика на тези остри епизоди, вкл. инсулти, от една страна, бърза [от няколко часа до няколко седмици] регресия на симптомите, от друга страна, склонност към рецидив); от страна на сетивните органи се откриват атрофия на зрителните нерви, пигментна ретинопатия и загуба на слуха.
Смята се, че следните механизми са важни в генезиса на IPE: [ 1 ] метаболитни нарушения в мозъка с развитие на лактатна ацидоза поради митохондриален енергиен дефицит; [ 2 ] церебрална исхемия, причинена от митохондриална ангиопатия на ниво малокалибрени артерии; [ 3 ] локално повишаване на невронната възбудимост поради митохондриална дисфункция в неврони, астроцити или капилярен ендотел, което постепенно се разпространява през мозъчната кора, се комбинира с развитието на оток и може да доведе до ламинарна некроза в церебралния кортекс.
На пръв поглед инсултът на MELAS изглежда като нормален инсулт, дължащ се на тромбоза или емболия. Всъщност подобните на инсулт епизоди при синдрома на MELAS са нетипични: те се срещат при млади хора, често се провокират от инфекциозни заболявания и могат да се появят под формата на мигреноподобно главоболие или конвулсивни атаки. ЯМР сканирането на остри PIE при синдрома на MELAS показва аномалии на усилване на сигнала в T2-претеглени или FLAIR (потискане на водата инверсия-възстановяване) изображения. Лезиите не съвпадат с басейните на големите церебрални артерии, но до голяма степен включват кората и подлежащото бяло вещество с умерено увреждане на дълбокото бяло вещество. Острите мозъчни лезии на ЯМР при синдром на MELAS могат да се променят, мигрират или дори да изчезнат ([ !!! ] се характеризира с флуктуация на огнищата, определени с ЯМР). Ангиографията разкрива липсата на тежка съдова патология: в допълнение към нормалните резултати може да се открие увеличение на калибъра на артериите, вените или капилярна хиперемия.
Невроморфологичните изследвания на мозъка при синдрома на MELAS показват наличието на мултифокална некроза, локализирана главно в мозъчната кора и субкортикалното бяло вещество, както и в малкия мозък, таламуса и базалните ганглии. Лезиите приличат на зони на инфаркт, но, както бе споменато по-горе, не съвпадат с басейните на големите церебрални съдове. Има също спонгиоформна дегенерация в мозъчната кора, капилярна пролиферация и изчерпване на невроните.
Помня! Подобните на инсулт епизоди при MELAS имат следните характеристики: [ 1 ] млада възраст (обикновено до 40 години); [ 2 ] честото наличие на провокиращ фактор (възникват след фебрилна температура, епилептичен пристъп, мигреноподобно главоболие); [ 3 ] любима локализация - тилна област; [ 4 ] огнищата, като правило, са разположени извън зоната на големите церебрални артерии, по-често разположени в кората или дълбоките структури на бялото вещество на мозъка.
При диференциалната диагноза на IPE и мозъчен инфаркт се вземат предвид следните симптоми:
■ постепенно, в продължение на няколко дни, увеличаване на фокалните неврологични симптоми (патофизиологичната основа за тази скорост на развитие е постепенното увеличаване на мозъчния енергиен дефицит поради нарушено окислително фосфорилиране в митохондриите);
■ постепенно намаляване на нивото на будност, което е в дисонанс с относително лек фокален неврологичен дефицит, не е придружено от вторичен стволов синдром и следователно не може да се обясни с увеличаване на мозъчния инфаркт и оток (основата на тези симптоми също е метаболитно разстройство, причинено от нарушение на енергийното снабдяване на мозъка);
■ развитие в острия период на повтарящи се локални и генерализирани епилептични припадъци, които според литературата се срещат при 2/3 от пациентите с IPE (припадъците не са свързани с нарушено мозъчно кръвообращение, тъй като източникът на тяхното генериране е активността на изхвърляне в двете полукълба на мозъка, а не в структури, ограничени до конкретен басейн от церебрални артерии; повтарящият се характер на гърчовете и липсата на изразени персистиращи фокални неврологични симптоми също не са характерни за острия мозъчен инфаркт);
■ пълна проходимост на мозъчните артерии според дуплексно сканиране на брахиоцефални артерии и церебрална ангиография, което не е характерно за исхемичния инсулт;
■ особености на невроизобразяващата картина: предимно кортикална локализация на огнищата и тяхното „задно разположение“, което е типично за MELAS и се обяснява с по-голямата уязвимост на невроните в тези области поради по-голямата им потребност от енергия; Друга характеристика на невроизображението е изчезването на някои огнища, които очевидно се основават на оток, а не на некроза на мозъчното вещество поради метаболитни нарушения.
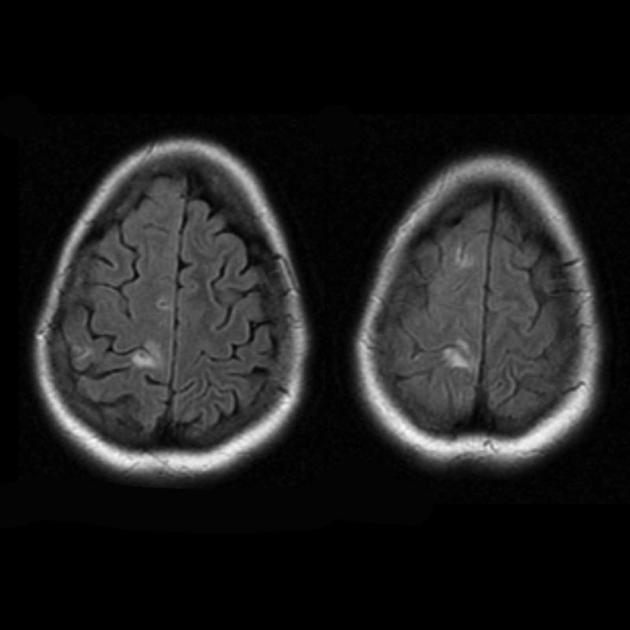
![]()
MELAS синдромна РАДИОПЕДИЯ.org
Една от основните прояви на синдрома MELAS също е мускулна слабост (миопатичен синдром). Въпреки това, неспецифичността на този симптом не позволява диагноза. Само когато се появят мигрена, конвулсии и/или удароподобни събития, може да се диагностицира началото на синдрома на MELAS.
Скрининговите тестове за синдрома MELAS са невроизобразяване и изследване на нивото [увеличаване] на лактат в кръвта (частично в цереброспиналната течност) - кръвен тест за съдържанието на млечна (лактат) и пирогроздена киселина (нивото на лактат в кръвта [норма] - венозна кръв - 0,5 - 2,2 mmol / l, артериална кръв - 0,5 - 1,6 mmol / l; съотношение лактат / пируват - 10/1). Диагнозата може да бъде потвърдена чрез ДНК изследване за определяне на най-честите мутации. При липса на общи точкови мутации при синдрома на MELAS, мускулната биопсия (която открива накъсани червени влакна [RKB] - миофибрили с високо съдържание на мутантен геном и голям брой пролифериращи променени митохондрии) може да помогне при диагностицирането. Също така (биопсия) ви позволява да определите наличието на биохимични дефекти в дихателната верига, свързани главно с ензимите сукцинат дехидрогеназа и цитохром оксидаза.

Лечението на синдрома MELAS включва две основни направления. Първата е постсиндромна терапия (основно внимание се обръща на епилепсия, захарен диабет и др.). Не се различава от конвенционалните подходи за лечение на синдроми. Облекчаването на епилептичните припадъци е необходимо, тъй като метаболитният стрес, който възниква по време на припадъци, може да провокира развитието на епизоди, подобни на инсулт. Производните на валпроевата киселина, широко използвани в епилептологията, инхибират митохондриалната функция и употребата им е нежелателна. Ако е невъзможно да отмените лекарството, трябва едновременно да приемате левокарнитин в доза до 100 mg / kg на ден. Фенитоинът и барбитуратите също трябва да се избягват. Второто направление на лечение е патогенетично, но в момента няма ефективна патогенетична терапия. Лечебната стратегия е насочена към подобряване на енергийния метаболизъм на клетката и включва назначаването на коензим Q или идебинон (нобен), препарати от янтарна киселина, витамини К1 и К3, никотинамид, рибофлавин, L-карнитин, антиоксиданти (мексидол, милдронат, витамини Е и С), лактат коректори ацидоза (димефосфон). [ !!!
] Необходимо е да се избягва употребата на лекарства, които потискат митохондриалната функция (барбитурати, валпроати, статини, глюкокортикоиди).
Прочетете повече за синдрома на MELAS в следните източници:
презентация "MELAS-синдром" Кузенкова Л.М., Глоба О.В.; Катедра по психоневрология, Изследователски институт по педиатрия, Научен център за детско здраве, Руската академия на медицинските науки, Москва [прочетете];
статия "Митохондриална енцефалопатия с удароподобни епизоди и лактатна ацидоза (синдром на MELAS): диагностични критерии, характеристики на епилептичните припадъци и подходи за лечение на примера на клиничен случай" Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A.; Държавна автономна институция на Ростовска област "Регионален консултативен и диагностичен център"; Катедрата по неврология и неврохирургия с курсове по мануална терапия и рефлексология на FPC и преподавателски състав на Федералната държавна бюджетна образователна институция за висше образование "Ростовски държавен медицински университет" на Министерството на здравеопазването на Русия (списание "Неврология, невропсихиатрия, психосоматика" № 9 (4), 2017) [прочети];
статия „Митохондриални цитопатии: синдроми MELAS и MIDD. Един генетичен дефект - различни клинични фенотипове” Муранова А.В., Строков И.А.; FGBOU VO "Първи Московски държавен медицински университет. ТЯХ. Сеченов" Министерство на здравеопазването на Руската федерация, Москва (Неврологичен журнал, № 1, 2017 г.) [прочетете];
статия "Инсулт-подобни епизоди при митохондриална енцефаломиопатия с лактатна ацидоза" L.A. Калашникова, Л.А. Добринина, А.В. Сахарова, Р.П. Чайковская, М.Ф. Мир-Касимов, Р.Н. Коновалов, А.А. Шабалина, М.В. Костирева, В.В. Гнездицки, С.В. Процки; Научен център по неврология на Руската академия на медицинските науки, Москва (списание "Анали по клинична и експериментална неврология" № 3, 2010) [прочетете];
статия „Неврологични нарушения при митохондриална енцефаломиопатия – лактатна ацидоза с удароподобни епизоди (MELAS синдром)“ D.A. Харламов, А.И. Крапивкин, В.С. Сухоруков, Л.А. Куфтина, О.С. Грознов; Московски изследователски институт по педиатрия и детска хирургия (списание "Руски бюлетин по перинатология и педиатрия" № 4 (2), 2012 г.) [прочетете];
статия "Инсултоподобен курс на митохондриална енцефаломиопатия (MELAS синдром)" I.N. Смирнова, Б.А. Кистенев, М.В. Кротенкова, З.А. Суслин; Научен център по неврология на Руската академия на медицинските науки, Москва (списание "Нервни болести" № 1, 2006 г.) [прочетете];
статия "Инсулти при митохондриални заболявания" N.V. Пизова, Катедра по нервни болести с курсове по неврохирургия и медицинска генетика, SBEE HPE "Ярославска държавна медицинска академия" (списание "Неврология, невропсихиатрия, психосоматика" № 9 (4), 2017) [прочетете];
статия "Исхемичен инсулт като проява на митохондриална енцефалопатия при млад пациент" Murzaliev A.M., Lutsenko I.L., Musabekova T.O., Akbalaeva B.A. (Списание "Наука и нови технологии" № 6, 2011) [прочетете];
статия „Епилепсия при синдром на MELAS“ Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А., Алиханов А.А., Рижков Б.Н., Петрухин А.С.; ГОУ ВПО РСМУ Росздрав; Руска детска клинична болница (Руски журнал за детска неврология "№ 3, 2009 г.) [прочетете];
статия "Алгоритъм за диагностика на митохондриални енцефаломиопатии" S.N. Илариошкин, Изследователски институт по неврология на Руската академия на медицинските науки (списание "Нервни болести" № 3, 2007 г.) [прочетете]
© Laesus De Liro
МЕЛАС (митохондриална енцефаломиопатия с лактатна ацидоза и инсулт-подобни епизоди, Английски MELAS е заболяване, което принадлежи към групата на митохондриалните заболявания. Митохондриите са органели, които имат собствена ДНК, която се предава и наследява по майчина линия. Типичните промени в изображенията включват инсулт-подобни огнища на различни етапи на развитие (модел на "променливо разпространение"), които не съвпадат с границите на артериалните съдови легла, има известно предразположение към появата на лезии в задните париетални и тилни лобове . MR спектроскопията може да покаже повишен пик на лактат дори при непроменен мозъчен модел.
Епидемиология
MELAS обикновено се проявява като епизоди, подобни на инсулт, при пациенти на възраст под 40 години при липса на рискови фактори за инсулт.
Клинична картина
МЕЛАСобикновено има рецидивиращо-ремитентно протичане, със или без прогресиращ дефицит. Клиничните прояви се характеризират с:
- общи прояви
- инсулт-подобни епизоди
- конвулсии
- лактатна ацидоза
- енцефалопатия
- деменция
- мускулна слабост
- глухота
Патология
Дефект под формата на точкова мутация на нуклеотид 3243 на митохондриална ДНК (A - G транслокация), кодираща левцинова тРНК, засяга дихателната верига (отговорна за производството на енергия). Синтезът на дефектен протеин води до смущения във функционирането на различни части на дихателната верига, което в крайна сметка води до изчерпване на NAD+ и NADH+. Това от своя страна води до превключване на метаболизма към анаеробна гликолиза, причинявайки натрупване на лактат, което прави кората на главния мозък по-податлива на хипоксия и в крайна сметка води до смърт на невроните. Тъй като някои митохондрии се прехвърлят с яйцеклетката, само част от митохондриите съдържат мутиралата ДНК. Тежестта на клиничните прояви зависи от броя на мутантните гени.
Диагностика
компютърна томография
- множество инфаркти
- калцификация на базалните ганглии
- по-изразени при по-възрастни пациенти
- атрофия
Магнитен резонанс
- хронични инфаркти
- множество съдови лезии
- може да бъде както симетричен, така и асиметричен
- по-чести са париетално-окципиталните и темпорално-теменните лезии
- остри инфаркти
- подуване на извивките с повишен MR сигнал на Т2 претеглени изображения
- възможно подобряване на контраста
- увреждане на субкортикалното бяло вещество
- повишен сигнал върху дифузионно претеглени изображения (Т2 трансилюминация) с незначителни (ако има) промени в DCI: промените в MELAS се характеризират с вазогенен, а не с цитотоксичен оток, както при инфаркт
MR спектроскопия: повишаване на пика на лактат е възможно дори при непокътнат мозъчен паренхим и цереброспинална течност.
Диференциална диагноза
- други митохондриални заболявания
- миоклонична епилепсия с разкъсани мускулни влакна
- Синдром на Kerns-Sayre
- епилептичен статус
- вирусен енцефалит
- церебрален васкулит
- Болест на Кройцфелд-Якоб
- инфаркт поради
- емболия
- дисекция
- CADASIL: неподкорови огнища
Ключови точки
Диагнозата MELAS се основава на следното: 1) удароподобни епизоди, настъпили преди 40-годишна възраст, 2) енцефалопатия с епилепсия и/или деменция, 3) наличие на лактатна ацидоза, увреждане на червени мускулни влакна и допълнителни критерии като повтарящи се главоболия, болка и повтарящо се повръщане. Типичните промени в изображенията включват подобни на инсулт огнища, калцификации на базалните ганглии и атрофия. Местоположението на повредата не съвпадащи с границите на артериалните съдови легла, както и възрастта и липсата на рискови фактори се противопоставят на инсулта. При синдрома на MELAS лезиите най-често се дължат на вазогенен оток и IBI обикновено не е намален или е по-слабо намален в сравнение със скорошен инфаркт.